

KATIE SIE, MADHAV SUBRAMANIAN, SERENA MCCALLA
ABSTRACT
Treatment refraction is a hallmark of small cell lung cancer (SCLC), occurring in almost 80% of patients after initial response to current treatment modalities. The aggressive nature and five-year survival rate of less than 5% of patients necessitates this research which identifies targetable genes to understand resistive mechanisms and mitigate poor prognosis. Public single-cell RNA sequencing (scRNA-Seq) datasets of untreated and DNA damage repair inhibitor (DDRi) treated samples were obtained through the Gene Expression Omnibus. Gene set enrichment analysis (GSEA) was performed to identify upregulated pathways associated with resistive mechanisms and prominent genes found in the leading edge subset of GSEA were visualized in RStudio and analyzed in Gene Expression Profiling Interactive Analysis 2 (GEPIA2) for survival impact. In this unsupervised study, the reactive oxygen species (ROS) and TGF-beta signaling pathways were two upregulated gene sets shared by both treatment types, supporting their association with resistive SCLC. Identified, shared leading edge subset genes of the ROS pathway included TXN, TXNRD1, NDUFB4, and LAMTOR5, which have been noted to allow cancerous cells to evade apoptosis and promote cell proliferation. Shared leading edge subset genes of the TGF-beta signaling pathway included HDAC1, CTNNB1, and SLC20A1, which have associated with the promotion of epithelial mesenchymal transitions, suppression of immune response, and increased tumor growth. This study identifies novel genes, previously unassociated with SCLC, that play a role in treatment refraction development in SCLC. Further experimentation may validate their potential as therapeutic targets to resensitize tumors and improve prognosis for individuals with resistant SCLC.
INTRODUCTION
Small Cell Lung Cancer: A Recalcitrant Cancer
Lung cancer currently causes the most cancer-related deaths in the United States, and consists of two major types: non-small cell lung cancer (NSCLC) and small cell lung cancer (SCLC; Siegel, Miller, & Jemal, 2020). The predominant cause of all lung cancers is smoking tobacco, but it is most closely associated with SCLC; 95% of SCLC cases occur in smokers. SCLC comprises approximately 15% of lung cancer cases and has a five-year survival rate of less than 5% (Sen, Gay, & Byers, 2018). The poor prognosis of SCLC is attributed to its quick proliferation and resistance to the limited treatment options available. Research to identify new therapies is hindered because SCLC is rarely identified before metastasis when surgical treatment is still an option, resulting in relatively few tissue samples to study (Yokouchi et al., 2020). Further experimentation is necessary to understand its resistive mechanisms and improve current treatment modalities.
Current Treatment Options for Small Cell Lung Cancer
Prescribed therapies for SCLC patients are largely dependent on the stage of the cancer at the time of diagnosis. Fewer than 30% of patients are diagnosed with limited stage SCLC, in which the tumor is confined to one lung. The most common treatment option for these patients is chemotherapy in combination with radiation or, less frequently, a lobectomy (Du et al., 2020; Saltos & Antonia, 2020). More than 70% of patients with SCLC are first diagnosed when the tumor has spread past the hemithorax, which is classified as the extensive stage of the disease (Lei et al., 2020). Patients first identified at this late stage of the disease often receive a standard treatment of platinum-based chemotherapy, such as cisplatin or carboplatin, in combination with etoposide as a first line of treatment to attempt tumor repression (Waqar & Morgensztern, 2017).
Additional treatments for SCLC attempt to exploit the high frequency of DNA mutations caused by smoking. In healthy tissue, Tumor Protein 53 (TP53) regulates the cell life cycle by promoting apoptosis in response to stress, preventing the rapid proliferation of cells as seen in cancer (Mantovani, Collavin, & Sal, 2019). Similarly, Retinoblastoma Protein 1 (RB1) reduces rapid cell division. Both TP53 and RB1 ensure arrest at the G1 and S phase to allow the cell to repair mutated DNA (Dyson, 2016). In SCLC, increased mutations in conjunction with the downregulation of TP53 and RB1 results in tumor cells with high dependence on DNA damage repair (DDR) mechanisms (Sen et al., 2018). Without these repair pathways, the tumoral DNA would be unstable, limiting tumor growth.
In SCLC tissue with abnormal DNA mutations, there is a higher dependence on poly-ADP-ribose polymerase (PARP) and checkpoint kinase 1 (CHK1), two proteins that regulate DDR. PARP without ADP ribose binds to sites of single strand DNA breaks (SSB). PARylation, the gathering of ADP ribose at the initial polymerase, then completes the PARP complex, which attracts other DNA repair enzymes, such as ligase III, polymerase beta, and X-ray repair cross-complementing protein 1 (XRCC1), to aid in the repair of DNA (Figure 1a) (Keung, Wu, & Vadgama, 2019). CHK1 also becomes an important DDR mechanism when TP53 and/or RB1 are downregulated (Figure 1c). Both PARP and CHK1 promote DDR in cancer tissues by stopping the cell cycle at the G2 or M phase, making these mechanisms necessary for tumor cell survival (Ferry et al., 2011). PARP and CHK1 are employed to repair the DNA in tumor cells and promote proliferation because the downregulation of P53 and RB1 allows tumors to avoid normal checkpoints, increasing the high mutational burden in SCLC.

Figure 1. Inhibition of DNA repair mechanisms. (a-b) Poly-ADP-ribose polymerase (PARP) a. The PARP polymerase is attracted to the site of the single strange break (SSB), which induces PARylation or the gathering of ADP ribose at the enzyme. The resulting complex then recruits other DNA repair enzymes that work with PARP to repair the SSB. b. The PARP inhibitor binds to a domain of the polymerase to prevent PARylation and the recruitment of other enzymes, evading DNA repair. PARP trapping, where the polymerase is tightly binding to the SSB, occurs until a double strand break forms. Apoptosis is a result of accumulating unrepaired DNA. ((Keung, Wu, & Vadgama, 2019) (c-d) Checkpoint Kinase 1 (CHK1) c. Ubiquitous loss of TP53 and RB1 prevents cell cycle from stopping at the G1 and S phase for DNA repair. CHK1 is necessary to stop cell cycle for DNA repair at the G2 or M phase. d. The CHK1 inhibitor deactivates CHK1. Cell cycle arrest never occurs, and DNA is not repaired, resulting in cell death. (Ferry et al., 2011)
As a result, DDR inhibitors (DDRi) are used as treatments to target specific DDR-associated proteins and were used to target cells in the datasets analyzed in this study (Figure 1b, d). PARP inhibitors (PARPi) bind to the initial polymerase and disrupt PARylation, resulting in PARP trapping, where the polymerase is unable to repair the DNA and remains bound to the SSB until a double strand break (DSB) forms (Figure 1b). The accumulation of DSBs causes apoptosis in tumor cells (Keung, Wu, & Vadgama, 2019). Similarly, CHK1 inhibitors (CHK1i) prevent DNA repair by disrupting cell cycle arrest at the G2 and M phase (Ferry et al., 2011). Unrepaired DNA eventually results in tumor cell death (Figure 1d).
Adaptive Resistance to Treatment
Initially, standard therapies induce responses in about 60% of patients, but all treated patients relapse (Stewart et al., 2020). The prognosis for retreatment of SCLC is dependent on the time between initial treatment and relapse. Patients who relapse after 60 days of initial chemotherapy doses are considered to be sensitive to treatment and can continue to receive standard therapies. Patients who relapse within 60 days are considered resistant (Asai, Ohkuni, Kaneko, Yamaguchi, & Akihito, 2014). Only 20% of resistant patients respond to current treatments; 80% require new treatment modalities to address initial treatment failures (Stewart et al., 2020).
There is reason to believe that treatment resistance may be a result of tumor heterogeneity. Variety in the individual tumor cells renders treatments ineffective when the therapeutic target is not present in a cell. As a result, cells lacking the therapeutic target will continue to proliferate. Treatments targeting PARP have been observed to be less efficient in the presence of homologous recombination, an alternative DNA repair pathway (Sen et al., 2018). Though PARPi can induce DNA damage, homologous recombination can repair DSBs, rendering the DDRi ineffective (Li & Heyer, 2008). Variety in the expression of vascular endothelial growth factor (VEGF) receptors have also been observed in SCLC. Drugs that target VEGF receptors initially delay angiogenesis, but cells without these receptors continue to proliferate and form new tumor blood vessels. The presence of other angiogenic stimulants like basic fibroblast growth factor makes VEGF an unsuitable target in SCLC (Kerbel, 2008). In a similar manner, heterogeneous expression of genes among different cell types also contribute to resistance. In a recent study analyzing xenografts of SCLC, varying transcriptional expression of genes involved in proliferation -- NOTCH pathway genes, MYC family genes, AURKA, AURKB, and ASCL1 -- was noted in treatment resistant tissue (Stewart et al., 2020).
Tumor heterogeneity is also present on the cellular level in relapsed SCLC, a result of upregulated epithelial mesenchymal transitions (EMTs). EMTs transform epithelial cells to mobilized mesenchymal stem cells (Cañadas et al., 2013). These mesenchymal stem cells and rare populations of cancer stem cells differentiate into various cell types, which move throughout the body as circulating tumor cells (CTCs) (Jin, Jin, & Kim, 2017; Kerbel, 2008). CTCs intravasate and extravasate blood vessels, further increasing treatment refraction as the stem cells metastasize and differentiate into different cell types (González-Silva, Quevedo, & Varela, 2020). This observed variety on the transcriptional and cellular level increases the difficulty of targeting and suppressing SCLC. Current treatments for SCLC are designed to treat the cancer in a homologous manner, and thus are of limited efficacy because cellular tumor heterogeneity frequently results in treatment refraction. Studying specific genes associated with such resistive mechanisms would allow individualized treatment for resistant patients based on their gene expression.
Objective: Rensensitizing Tumors to Treatment
Researchers have conducted clinical trials to test drugs with potential resensitizing properties. Topotecan has been used as a second line of treatment after resistance, though a median response period of less than four months and adverse side effects including weakened immune system and anemia have encouraged further research for other treatments (Ready et al., 2019). In August of 2018, the FDA-approved immunotherapy, nivolumab, was reported to have a response rate of about 10% in resistant SCLC tissue. A combination of nivolumab and another immunotherapy, ipilimumab, provided a high response rate, but the clinical trial was discontinued after cytotoxicity caused fatal outcomes. Another immunotherapy, pembrolizumab, was approved in June of 2019 by the FDA with a 19.3% response rate. Both nivolumab and pembrolizumab did not significantly improve overall survival in patients, demanding additional studies on treatment refraction as efforts to resensitize the majority of resistant tumors to treatment have been unsuccessful and survival rates remain low (Saltos & Antonia, 2020; Sen, Gay, & Byers, 2018). Current DDRi treatments become less effective after relapse, necessitating further research on mechanisms driving treatment refraction.
Thus, this study aims to identify novel genes that can target resistant SCLC cells, further the understanding of SCLC resistive mechanisms, and lead to the improvement of SCLC treatments through analyzing scRNA Seq datasets. In recent years, the Gene Expression Omnibus (GEO), which is supported by the National Center for Biotechnology Information (NCBI), has provided a repository for scientists to publicly share sequenced data. Datasets of untreated CTC-derived xenografts as well as DDRi treated xenografts were evaluated and analyzed in this study. These datasets included cells treated with PARPi talazoparib and CHK1i prexasertib. Untreated, which served as the control group, and treated datasets were analyzed in this study to discern significantly upregulated genes and pathways in resistive SCLC cells from upregulated biomarkers in SCLC cells that have not been treated.
The researchers who deposited these datasets have not performed deep analysis of these datasets obtained through scRNA Seq (Stewart et al., 2020). In contrast to bulk RNA sequencing, scRNA Seq allows the in depth analysis of gene expression of individual cells and comparisons between cells (Leucken & Theis, 2019; Hwang, Lee, & Bang, 2018). The complexity of scRNA Seq has provided this study with a comprehensive understanding of heterogeneous, DDRi-treated SCLC samples through upregulated pathways identified in GSEA and differentially expressed genes identified in R Studio and studied using Gene Expression Profiling Interactive Analysis 2 (GEPIA2) (http://gepia2.cancer-pku.cn).
Further insight into mechanisms driving the development of treatment refraction through this study’s analyses can identify genes associated with tumor growth in resistive SCLC. Through future experimentation, identified genes can be downregulated to verify their role in treatment resistance and possibly be targeted in future medication to improve treatments for resistant SCLC patients
METHODS
Data Acquisition
The current study used data solely from the Stewart et al. 2020 study obtained through the GEO repository (https://www.ncbi.nlm.nih.gov/geo/). In their study, Stewart et al. injected CTC-derived xenografts (CDXs) into the flank of mice and resected them after reaching the threshold volume (n = 500-800mm3). Stewart et al. then dissociated the CDXs from the mice to isolate human cells, and scRNA Seq was performed. This study utilized the datasets from the Stewart et al. 2020 study and analyzed all data using a MacBook Pro.
Although three of each differentially, DDRi-treated CDX samples were available, all datasets were not analyzed due to the limited processing power of the MacBook Pro used in this study. The limitations of the data available as well as the tools used for analyses are considered under the limitations section. Untreated CDX samples (n = 2), talazoparib-treated CDX samples (n = 2), and prexasertib-treated CDX samples (n = 1), all of which were derived from patient SC4, were analyzed. Only datasets from patient SC4 were treated with DDRi and, thus, used in this study. Patient SC4 received one cycle of platinum-based chemotherapy and was concluded to be sensitive to treatment. Datasets were chosen to represent the SCLC tumors before and after treatment refraction to DDRi.
Quality Control
In this study, RStudio (V. 1.3.959) was used to conduct quality control of datasets, cluster data, and visualize differentially expressed genes. The ggplot2 and cowplot packages were used in conjunction to create figures from the data. The Seurat and dplyr packages were used analysis of the scRNA Seq datasets, allowing for manipulating complex data, clustering cells, and quality control.
Multiple steps were first taken using the Seurat, ggplot2, cowplot, and dplyr packages in RStudio to ensure the presence of only high quality cells in the analysis of the datasets. Cells with fewer than 200 genes and genes expressed in fewer than three cells were not analyzed. The percentage of mitochondrial DNA (mtDNA) and number of RNA counts of each cell were visualized in a violin plot. High expression of mtDNA, which is generally seen when a nucleus has ruptured, suggests poor quality and dying cells, which should not be included in the analysis of the dataset. High RNA counts also suggest the presence of sampling noise from doublets or cells that represent the transcriptome of two cells. Thresholds were independently chosen for each dataset based on the distribution seen in the violin plot (Table 1).

Table 1. Quality control thresholds for each dataset. The percentage of mitochondrial DNA (mtDNA) and number of RNA counts were accounted for when classifying poor quality cells that would be excluded from analysis in this study. Cells remaining after removing poor quality cells were classified as high quality cells and are defined by the thresholds shown.
Data Clustering
Data clustering was performed in RStudio using the Seurat, ggplot2, cowplot, and dplyr packages to identify various cell clusters involved in different biological processes. Untreated samples GSM4104153 and GSM4104154 were integrated with talazoparib-treated samples GSM4104160 and GSM4104151 for analysis of pathways and genes upregulated in resistant PARPi SCLC cells. Untreated sample GSM4104153 and prexasertib-treated sample GSM4104158 were integrated for analysis of pathways and genes upregulated in resistant CHK1i SCLC cells. Equal amounts of untreated and treated samples were integrated to ensure similar sample sizes.
Principal component analysis was performed on both integrated datasets to identify the 40 most differential dimensions. Dimensionality reduction was necessary to present the data in two-dimensional plots. An elbow plot was constructed to demonstrate the variance of each principal component. The first 35 dimensions in both integrated datasets were the most differential relative to one another and chosen for further analysis. A uniform manifold approximation and projection (UMAP) plot was then constructed to cluster similar cells and visualize the heterogeneity in the SCLC tissue.
Gene Set Enrichment Analysis (GSEA)
GSEA was performed to identify enriched pathways that may play a role in treatment refraction. The hallmark gene sets from the Molecular Signatures Database (MSigDB) were used to identify pathways in the datasets for all GSEA. Phenotypic permutations (n = 1000) were used in all analyses to increase the precision of calculated p values. The remaining parameters for GSEA were set to default: weighted enrichment statistic, Signal2Noise metric for ranking genes, and real gene list sorting mode. Enrichment scores (ES) were calculated through a running-sum statistic that uses a ranked list of genes from the loaded dataset and increases when a gene from the list is present in the pathway being analyzed. Normalized enrichment scores (NES) consider the testing of multiple hypotheses and were used in this study (Subramanian et al., 2005). Statistical significance of NES was determined by the p value (p < 0.001), false discovery rate (FDR < 0.001), and familywise-error rate (FWER < 0.005).
GSEA was performed on each cluster of the talazoparib-integrated and prexasertib-integrated datasets to identify upregulated gene sets and better understand the various biological processes present. GSEA was also performed to identify upregulated gene sets in treated compared to untreated cells.
Leading Edge Subset Analysis
Common genes contributing to shared GSEA enriched pathways between treatments were identified and further analyzed for their role in SCLC treatment refraction. Leading edge subset genes were further presented in dot plots and violin plots using Seurat, dyplr, ggplot2, and cowplot packages in RStudio to demonstrate differential expression between treatment conditions.
Survival analysis was also performed on each identified gene using GEPIA2. As established by Tang, Kang, Li, Chen, & Zhang in 2019, GEPIA2 runs the Mantel-Cox test and determines the survival effects of the high and low expression of selected genes using information from The Cancer Genome Atlas (TCGA) and Genotype-Tissue Expression project. Databases used in GEPIA2 do not include SCLC data, but survival analysis was still performed to determine the significance (p < 0.05) of identified genes on the survival rates in other cancers.
RESULTS
Data Clustering: Identifying Heterogeneous Clusters
Dimensionality reduction and clustering was performed and portrayed on a UMAP plot to visualize the heterogeneity in the talazoparib and prexasertib-integrated samples. Seven clusters were identified in each integrated dataset as displayed by the variously colored clusters in Figure 2. GSEA was then performed to examine the gene sets uniquely present in individual clusters of both integrated datasets. Cluster 1 of the talazoparib-integrated dataset and cluster 2 of the prexasertib-integrated dataset shared three hallmark gene sets: both MYC targets and oxidative phosphorylation. Cluster 3 of the talazoparib-integrated dataset and cluster 0 of the prexasertib-integrated dataset shared two hallmark gene sets: apoptosis and inflammatory response.

Figure 2. Heterogeneous clusters. a) UMAP plot of the talazoparib-integrated dataset. b) UMAP plot of prexasertib-integrated dataset. a-b) Presence of 7 individual clusters in both UMAP plots suggest tumoral heterogeneity. c) Bar graph representing enriched gene sets in cluster 1 of the talazoparib-integrated dataset. d) Bar graph representing enriched gene sets in cluster 2 of the prexasertib-integrated dataset. c-d) MYC genes and oxidative phosphorylation were upregulated in one cluster of each treatment type, suggesting treatment resistance. e) Bar graph representing enriched gene sets in cluster 3 of talazoparib-integrated dataset. f) Bar graph representing enriched gene sets in cluster 0 of prexasertib-integrated dataset. e-f) Apoptosis and inflammatory response were upregulated in one cluster of each treatment types, implying successful treatment response. (p < 0.001, FDR < 0,001, FWER < 0.005)
Gene Set Enrichment Anaylsis (GSEA)
GSEA was performed to examine the gene sets enriched in the treated condition versus the untreated condition of both integrated datasets (Figure 3). The reactive oxygen species (ROS) pathway and TGF-beta signaling gene set were upregulated in both treatment types as shown by the green line skewed to the right in Figure 3c and 3d (p < 0.001, FDR < 0.001, FWER <0.005).

Figure 3. Enriched GSEA pathways shared between talazoparib and prexasertib datasets. a) Bar graph of the top ten upregulated gene sets in the talazoparib-treated tumor cells. b) Bar graph of the top ten upregulated gene sets in the prexasertib-treated tumor cells. a-b) Enrichment of various biological pathways indicates heterogeneity in the SCLC environment (p < 0.001, FDR < 0.001, FWER < 0.005). c) Enrichment plot of the ROS and TGF-beta signaling pathways in talazoparib-treated cells from GSEA. d) Enrichment plot of the ROS and TGF-beta signaling pathways in prexasertib-treated cells from GSEA. c-d) Enrichment plots are heavily skewed to the left, indicating upregulation in the treated conditions.
Leading Edge Subset Analysis
The leading edge subset genes contributing to the upregulation of the ROS pathway of both talazoparib-treated and prexasertib-treated datasets were examined in RStudio for differential expression. Four differentially-expressed, shared genes between both treatment types were identified: TXN, TXNRD1, NDUFB4, and LAMTOR5 (Figure 4). All common genes were studied in GEPIA2 for their impact on survival, but downregulation of TXN was the only gene to show significant (p < 0.05) survival benefit in lung adenocarcinoma (LUAD) and lung squamous cell carcinoma (LUSC).
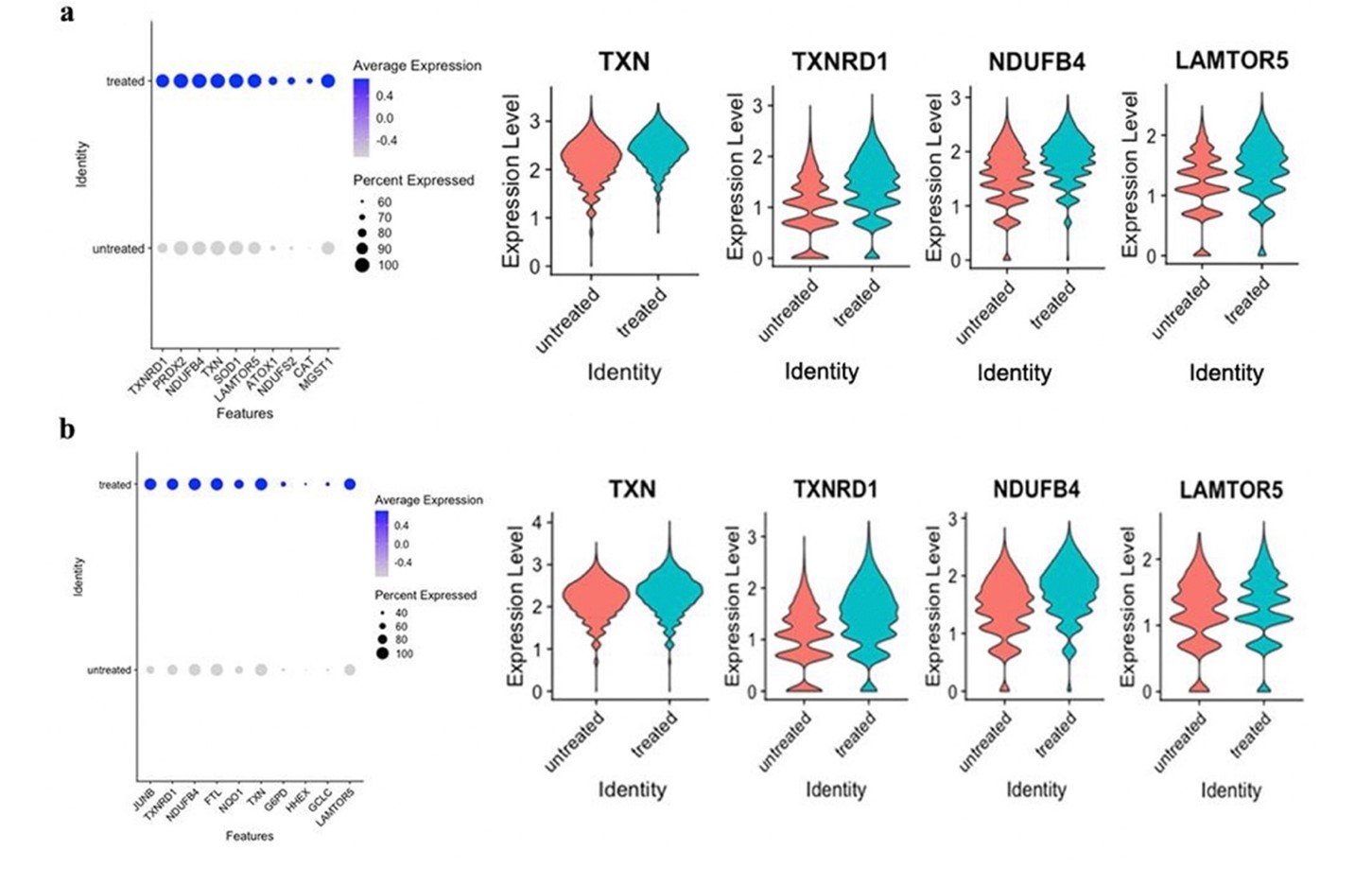
Figure 4. Differential expression of leading edge subset genes associated with the reactive oxygen species pathway. a) Dot plot of the first ten genes in the leading edge subset of the ROS pathway in the talazoparib-treated samples, and violin plots of TXN, TXNRD1, NDUFB4, and LAMTOR5. b) Dot plot of the first ten genes in the leading edge subset of the ROS pathway in the prexasertib-treated samples, and violin plots of TXN, TXNRD1, NDUFB4, and LAMTOR5. a-b) TXN, TXNRD1, NDUFB4, and LAMTOR5 were common, highly contributive genes in the ROS pathways of both treatment types.
The leading edge subset genes contributing to the upregulation of the TGF-beta signaling pathway of both talazoparib and prexasertib datasets were also examined in RStudio for differential expression (Figure 5). Three shared genes between both treatment types were identified: HDAC1, CTNNB1, and SLC20A1.
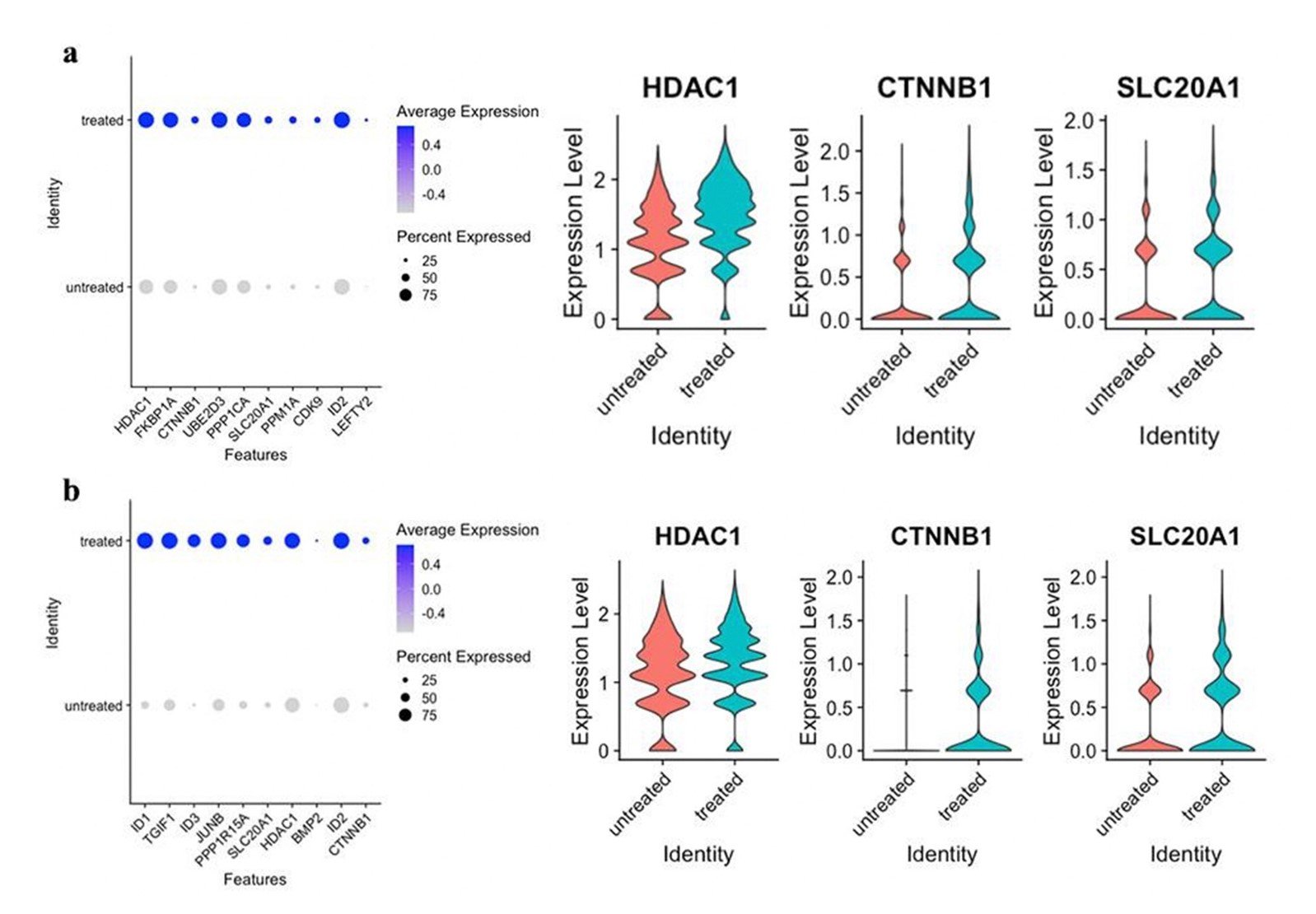
Figure 5. Differential expression of leading edge subset genes associated with the TGF-beta signaling pathway. a) Dot plot of the first 10 genes in the leading edge subset of the TGF-beta signaling pathway in the talazoparib-treated samples, and violin plots of HDAC1, CTNNB1, and SLC20A1. b) Dot plot of the first 10 genes in the leading edge subset of the TGF-beta signaling pathway in the prexasertib-treated samples, and violin plots of HDAC1, CTNNB1, and SLC20A1. a-b) HDAC1, CTNNB1, and SLC20A1 were common, highly contributive genes in the TGF-beta signaling of both treatment types.
DISCUSSION
This study identified genes previously unassociated with SCLC that may contribute to the development of resistance in DDRi-treated SCLC through bioinformatic analyses of upregulated pathways in resistant tumor cells. Public datasets were analyzed with no intended results but with the purpose of furthering current understanding of resistant SCLC.
Identifying Heterogeneous Clusters
Seven clusters were identified in each integrated dataset, suggesting the presence of heterogeneous biological processes as cells were clustered based on similar gene expression (Figure 2; Shue et al., 2018). The enriched gene sets present in the GSEA of individual clusters demonstrate heterogeneity in treatment response. The upregulation of MYC oncogenes, which have been correlated to cell proliferation, in one individual cluster from both integrated dataset implies treatment resistance (Bottger et al., 2020). Oxidative phosphorylation enrichment further supports treatment refractory by suggesting increased glycolysis to support tumor growth (Rodriguez-Enriquez, 2019). In contrast, the shared upregulation of apoptosis and inflammatory response indicates some successful treatment response (Spigel and Socinski, 2013). Indications of treatment resistance and response in various individual clusters of both talazoparib-integrated and prexasertib-integrated datasets support current understandings of the heterogeneity in SCLC.
Common GSEA Results Between Talazoparib and Prexasertib Datasets
The ROS and TGF-beta signaling gene sets were upregulated in both treatment types (p < 0.001, FDR < 0.001, FWER <0.005). In the tumor environment, the ROS pathway has been associated with epidermal growth factor receptors, promoting proliferation, differentiation, and metastasis, but excessive ROS can also cause cell death. (Weng et al., 2018). The TGF-beta signaling pathway has also been correlated with tumor survival through increased migration and EMTs and tumor suppression through apoptosis (Colak and Dijke, 2017). Current understanding of the ROS and TGF-beta signaling pathways support the plausibility of their relation to the development of resistance in SCLC.
Shared Leading Edge Subset Genes
Reactive Oxygen Species (ROS) Pathway
There were four leading edge subset genes contributing to the upregulation of the ROS pathway of both talazoparib-treated and prexasertib-treated datasets: TXN, TXNRD1, NDUFB4, and LAMTOR5 (Figure 4). Upregulation of these four common genes may be a result of oxidative stress as has been noted in previous research (Zhang et al., 2019; Zhu et al., 2019; O’Malley et al., 2020). TXN and TXNRD1 maintain reductive pathways when imbalanced amounts of free radicals disrupt homeostasis. Previous studies have correlated high expression of TXN and TXNRD1 with poor overall survival in breast, pancreatic, head, neck, prostate, and colon cancers (Stafford et al., 2018). Upregulation of TXN and TXNRD1 has also been observed to increase resistivity to cisplatin chemotherapy in NSCLC (Zhu et al., 2019). The NDUFB4 gene codes for a NADH dehydrogenase, which is involved in mitochondrial activities. Increased production of ROS and mitochondrial dysfunction results in upregulated NDUFB4 and evasion of apoptosis (O’Malley et al., 2020; Ghosh & Girigoswmani, 2008). LAMTOR5 translates a protein that interacts with transcription factor NF-kB to induce cell proliferation through mitogen-activated protein kinase (MAPK) and mammalian target of rapamycin (mTOR) (Zhang et al., 2019). Past research has correlated increased LAMTOR5 expression with the activation of GLUT1 in liver cancer, increasing glucose metabolism to support tumor growth (Zhou et al., 2019).

Figure 6. Survival analysis of TXN in lung adenocarcinoma and lung squamous cell carcinoma. Upregulation of TXN in the high TXN group shows a significant, negative impact on survival outcome in lung adenocarcinoma and lung squamous cell carcinoma compared to the low TXN group. Although previous studies have not correlated upregulated TXN to the survival outcome of SCLC, this analysis supports the possibility that there exists a relationship. Analysis was performed using GEPIA2. (p < 0.03)
Survival analysis was also performed in GEPIA2 to determine the role of identified genes in the context of other cancers. Downregulation of TXN was the only gene to show significant (p < 0.05) survival benefit in lung adenocarcinoma (LUAD) and lung squamous cell carcinoma (LUSC) (Figure 6). Current understanding of TXN, TXNRD1, NDUFB4, and LAMTOR5 in cancer environments in combination with its prominent presence in the ROS pathway of treated samples here suggest a notable role of these identified genes in resistance against treatment.
TGF-Beta Signaling
There were three leading edge subset genes contributing to the upregulation of the TGF-beta signaling pathway of both talazoparib and prexasertib datasets: HDAC1, CTNNB1, and SLC20A1 (Figure 5). Upregulation of HDAC1, CTNNB1, and SLC20A1 induce different aspects of the TGF-beta signaling pathway to promote cell growth and treatment resistance. HDAC1 is a histone deacetylase that collaborates with the acetylation of histone 3 to promote EMTs, increasing tumoral heterogeneity and metastasis (Qiao et al., 2020). Upregulation of HDAC1 has also been noted to prevent the transcription of apoptosis stimulating protein of p53-2 (ASPP2), fostering cell proliferation (Li et al., 2018 A). CTNNB1 produces β-catenin which regulates cell adhesion and suppression of immune responses in cancers (Montepreville et al., 2020). Overexpression of CTNNB1 has also been reported to worsen overall survival in acute myeloid leukemia, colorectal, breast, and liver cancer by increased cell differentiation and inhibition of apoptosis (Li et al., 2018 B). SLC20A1 induces upregulation of NCAD, VEGF, and VIM, which are genes known for promoting tumor growth through angiogenesis. Increased expression of SLC20A1 has also been correlated with increased tumor size, decreased tumor necrosis factor-induced apoptosis, and worse survival in breast cancer (Guo and Wang, 2008; Li et al., 2019). The known function of HDAC1, CTNNB1, and SLC20A1 as well as their prominence in the TGF-beta signaling pathway of talazoparib and prexasertib-treated datasets in this study support their potential role as biomarkers for treatment resistance in SCLC.
Limitations
Several limitations were present in conducting this study. Datasets used from the Stewart et. al 2020 study were all derived from CDXs created with CTCs of one patient, which renders the concern of generalizability of the results to a larger population. Although this limitation was controlled with strict thresholds for statistical significance, future studies should include samples from a larger population of individuals. The limited processing power of the Macbook Pro used for analyses in this study was also a restricting factor as not all available datasets could be analyzed. The results could have been altered if all available datasets were analyzed. Future studies should utilize more data sets and a device with the ability to analyze all data sets desired.
Future Investigations
The role of identified genes in DDRi-resistant SCLC can be further validated in future investigations with therapeutic targets. TXN and TXNRD1 can be downregulated using FDA-approved arsenic trioxide, which is in current clinical trials for leukemia, basal cell cancer, and neuroblastoma (Arnér, 2019). Auranofin, another drug targeting TXN and TXNRD1, is in clinical trials for ovarian cancer, NSCLC, and SCLC to inhibit PKCι and mTOR (Ross & Lou, 2020). HDAC1 can be targeted using FDA-approved vorinostat, which has been tested in renal cell carcinoma (Li et al., 2018 A; Richon, 2006). NDUFB4, LAMTOR5, CTNNB1, and SLC20A1 can be targeted using clustered regularly interspaced short palindromic repeats (CRISPR) and the Cas9 protein. Engineered guide RNAs can be used to target and downregulate the desired gene (Jeong et al., 2020; Xu et al., 2018). The knockout of genes established in this study should also be examined in SCLC resistant tumors treated with other therapies. Heterogeneity in SCLC promotes a variety of resistive mechanisms as a response to specific treatments. The role of TXN, TXNRD1, NDUFB4, LAMTOR5, CTNNB1, and SLC20A1 should be investigated in differentially treated tumors to understand their potential as resensitizing therapeutic targets (Stewart et al., 2020).
Conclusion
Using bioinformatic analyses, this study identified ROS and the TGF-beta signaling pathways as well as specific genes previously unassociated with resistant SCLC as contributors to SCLC treatment resistance. TXN, TXNRD1, NDUFB4, and LAMTOR5 are upregulated in the presence of oxidative stress, which may have been a result of treatment response to the DDRi, and inadvertently increase treatment refractory through cell proliferation. Upregulation of HDAC1, CTNNB1, and SLC20A1 in the TGF-beta signaling pathway may also indicate resistance to treatment through increased EMTs, inhibition of apoptosis, and suppressed immune response. These identified genes may serve as therapeutic targets in future treatments as standard treatment for SCLC has not improved in recent decades and median survival after diagnosis is less than two years (Mohan et al., 2020; Koinis, Kotsakis, & Georgoulias, 2016). Although additional exploration is required, this study provides a further understanding of the resistive mechanisms of SCLC and has the potential to improve treatments as well as prognosis for people with resistant SCLC.
REFERENCES
Arnér, E. S. (2019). Perspectives of TrxR1-based cancer therapies. In H. Sies (Ed.), Oxidative Stress: Eustress and Distress, 1st ed., pp. 639-667, Amsterdam, Netherlands: Elsevier. available: doi:10.1016/B978-0-12-818606-0.00031-6
Böttger, F., Semenova, E. A., Song, J., Ferone, G., Vliet, J. V., Cozijnsen, M., Bhaskaran, R., Bombardelli, L., Piersma, S. R., Pham, T. V., Jimenez, C. R., & Berns, A. (2019). Tumor heterogeneity underlies differential cisplatin sensitivity in mouse models of small-cell lung cancer. Cell Reports, 27(11), available: doi:10.1016/j.celrep.2019.05.057
Butler, A., Hoffman, P., Smibert, P., Papalexi, E., & Satija, R. (2018). Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nature Biotechnology, 36(5), 411-420, available: doi:10.1038/nbt.4096
Cañadas, I., Rojo, F., Taus, A., Arpi, O., Arumi-Uria, M., Pijuan, L., Menéndez, S., Zazo, S., Dómine, M., Salido, M., Mojal, S., Garcia de Herreros, A., Rovira, A., Albanell, J., & Arriola, E. (2013). Targeting epithelial-to-mesenchymal transition with met inhibitors reverts chemoresistance in small cell lung cancer. Clinical Cancer Research, 20(4), 938-950, available: doi:10.1158/1078-0432.ccr-13-1330
Colak, S., & Dijke, P. T. (2017). Targeting TGF-β Signaling in Cancer. Trends in Cancer, 3(1), 56-71, available: doi:10.1016/j.trecan.2016.11.008
Conesa, A., Madrigal, P., Tarazona, S., Gomez-Cabrero, D., Cervera, A., Mcpherson, A., Szczesniak, M. W., Gaffney, D. J., Elo, L. L., Zhang, X., & Mortazavi, A. (2016). Erratum to: A survey of best practices for RNA-seq data analysis. Genome Biology, 17(1), available: doi:10.1186/s13059-016-1047-4
Du, J., Li, Y., Wang, L., Zhou, Y., Shen, Y., Xu, F., & Chen, Y. (2020). Selective application of neuroendocrine markers in the diagnosis and treatment of small cell lung cancer. Clinica Chimica Acta, 509, 295-303, available: doi:10.1016/j.cca.2020.06.037
Ferry, G., Studeny, A., Bossard, C., Kubara, P. M., Zeyer, D., Renaud, J., Casara, P., Nanteuil, G., Wierzbicki, M., Pfeiffer, B., Prudhomme, M., Leonce, S., Pierré, A., Boutin, J. A., & Golsteyn, R. M. (2011). Characterization of novel checkpoint kinase 1 inhibitors by in vitro assays and in human cancer cells treated with topoisomerase inhibitors. Elsevier, 89, 259-268), available: doi:10.1016/j.lfs.2011.06.010
Ghosh, R., & Girigoswami, K. (2008). NADH dehydrogenase subunits are overexpressed in cells exposed repeatedly to H2O2. Mutation Research/Fundamental and Molecular Mechanisms of Mutagenesis, 638(1-2), 210-215), available: doi:10.1016/j.mrfmmm.2007.08.008
González-Silva, L., Quevedo, L., & Varela, I. (2020). Tumor functional heterogeneity unraveled by scRNA-seq technologies. Trends in Cancer, 6(1), 13-19), available: doi:10.1016/j.trecan.2019.11.010
Guo, X., & Wang, X. (2008). Signaling cross-talk between TGF-β/BMP and other pathways. Cell Research, 19(1), 71-88, available: doi:10.1038/cr.2008.302
Hwang, B., Lee, J. H., & Bang, D. (2018). Single-cell RNA sequencing technologies and bioinformatics pipelines. Experimental & Molecular Medicine, 50(8) ), available: doi:10.1038/s12276-018-0071-8
Jeong, C., Kang, H., Hong, S., Byeon, E., Lee, J., Lee, Y. H., Choi, I., Bae, S., & Lee, J. (2020). Generation of albino via SLC45a2 gene targeting by CRISPR/Cas9 in the marine medaka Oryzias melastigma. Marine Pollution Bulletin, 154, 111038, available: doi:10.1016/j.marpolbul.2020.111038
Kerbel, R. S. (2008). Tumor angiogenesis. New England Journal of Medicine, 358(19), 2039-2049, available: doi:10.1056/nejmra0706596
Keung, M., Wu, Y., & Vadgama, J. (2019). PARP Inhibitors as a Therapeutic Agent for Homologous Recombination Deficiency in Breast Cancers. Journal of Clinical Medicine, 8(4), 435, available: doi:10.3390/jcm8040435
Koinis, F., Kotsakis, A., & Georgoulias, V. (2016). Small cell lung cancer (SCLC): no treatment advances in recent years. Translational lung cancer research, 5(1), 39–50, available: https://doi.org/10.3978/j.issn.2218-6751.2016.01.03
Lei, Y., Feng, H., Qiang, H., Shang, Z., Chang, Q., Qian, J., Zhang, Y., Zhong, R., Fan, X., & Chu, T. (2020). Clinical characteristics and prognostic factors of surgically resected combined small cell lung cancer: A retrospective study. Lung Cancer, 146, 244-251, available: doi:10.1016/j.lungcan.2020.06.021
Li, J., Dong, W., Li, Z., Wang, H., Gao, H., & Zhang, Y. (2019). Impact of SLC20A1 on the Wnt/β‑catenin signaling pathway in somatotroph adenomas. Molecular Medicine Reports, 20, 3276-3284, available: doi:10.3892/mmr.2019.10555
Li, H., Wang, X., Zhang, C., Cheng, Y., Yu, M., Zhao, K., Ge, W., Cai, A., Zhang, Y., Han, F., & Hu, Y. (2018). HDAC1-induced epigenetic silencing of ASPP2 promotes cell motility, tumour growth and drug resistance in renal cell carcinoma. Cancer Letters, 432, 121-131, available: doi:10.1016/j.canlet.2018.06.009
Li, X., Guo, H., Zhou, J., Wu, D., Ma, J., Wen, X., Zhang, W., Xu, Z., Lin, J., & Jun, Q. (2018). Overexpression of CTNNB1 : Clinical implication in Chinese de novo acute myeloid leukemia. Pathology - Research and Practice, 214(3), 361-367, available: doi:10.1016/j.prp.2018.01.003
Luecken, M. D., & Theis, F. J. (2019). Current best practices in single‐cell RNA‐seq analysis: A tutorial. Molecular Systems Biology, 15(6), available: doi:10.15252/msb.20188746
Mohan, S., Foy, V., Ayub, M., Leong, H. S., Schofield, P., Sahoo, S., Descamps, T., Kilerci, B., Smith, N. K., Carter, M., Priest, L., Zhou, C., Carr, T. H., Miller, C., Faivre-Finn, C., Blackhall, F., Rothwell, D. G., Dive, C., & Brady, G. (2020). Profiling of Circulating Free DNA Using Targeted and Genome-wide Sequencing in Patients with SCLC. Journal of Thoracic Oncology, 15(2), 216-230, available: doi:10.1016/j.jtho.2019.10.007
Montpréville, V. T., Lacroix, L., Rouleau, E., Mamodaly, M., Leclerc, J., Tutuianu, L., Planchard, D., Boulate, D., Mercier, O., Besse, B., Fadel, E., & Ghigna, M. (2020). Non-small cell lung carcinomas with CTNNB1 (beta-catenin) mutations: A clinicopathological study of 26 cases. Annals of Diagnostic Pathology, 46, 151522, available: doi:10.1016/j.anndiagpath.2020.151522
O’Malley, J., Kumar, R., Inigo, J., Yadava, N., & Chandra, D. (2020). Mitochondrial Stress Response and Cancer. Trends in Cancer, 6(8), 688-701, available: doi:10.1016/j.trecan.2020.04.009
Qiao, Y., Wang, Z., Tan, F., Chen, J., Lin, J., Yang, J., Hui, L., Wang, X., Sali, A., Zhang, L., & Zhong, G. (2020). Enhancer Reprogramming within Pre-existing Topologically Associated Domains Promotes TGF-β-Induced EMT and Cancer Metastasis. Molecular Therapy, available: doi:10.1016/j.ymthe.2020.05.026
Ready, N., Farago, A. F., Braud, F. D., Atmaca, A., Hellmann, M. D., Schneider, J. G., Spigel, D. R., Moreno, V., Chau, I., Hann, C. L., Eder, J. P., Steele, N. L., Pieters, A., Fairchild, J., & Antonia, S. J. (2019). Third-Line Nivolumab Monotherapy in Recurrent SCLC: CheckMate 032. Journal of Thoracic Oncology, 14(2), 237-244, available: doi:10.1016/j.jtho.2018.10.003
Richon, V. M. (2006). Cancer biology: Mechanism of antitumour action of vorinostat (suberoylanilide hydroxamic acid), a novel histone deacetylase inhibitor. British Journal of Cancer, 95(S1), 2-6, available: doi:10.1038/sj.bjc.6603463
Rodríguez-Enríquez, S., Pacheco-Velázquez, S. C., Marín-Hernández, Á, Gallardo-Pérez, J. C., Robledo-Cadena, D. X., Hernández-Reséndiz, I., García-García, J. D., Belmont-Díaz, J., Lópex-Marure, R., Hernández-Esquivel, L., Sánchez-Thomas, R., & Moreno-Sánchez, R. (2019). Resveratrol inhibits cancer cell proliferation by impairing oxidative phosphorylation and inducing oxidative stress. Toxicology and Applied Pharmacology, 370, 65-77, available: doi:10.1016/j.taap.2019.03.008
Ross, H., & Lou, Y. (2020, April 23). PKCι & mTOR Inhibition With Auranofin+Sirolimus for Squamous Cell Lung Cancer. Retrieved August 02, 2020, from https://www.mayo.edu/research/clinical-trials/cls-20115754
Saltos, A., & Antonia, S. (2020). Breaking the impasse. Clinics in Chest Medicine, 41(2), 269-280, available: doi:10.1016/j.ccm.2020.02.011
Sen, T., Gay, C. M., & Byers, L. A. (2018). Targeting DNA damage repair in small cell lung cancer and the biomarker landscape. Translational Lung Cancer Research, 7(1), 50-68, available: doi:10.21037/tlcr.2018.02.03
Shue, Y. T., Lim, J. S., & Sage, J. (2018). Tumor heterogeneity in small cell lung cancer defined and investigated in pre-clinical mouse models. Translational Lung Cancer Research, 7(1), 21-31, available: doi:10.21037/tlcr.2018.01.15
Siegel, R. L., Miller, K. D., & Jemal, A. (2020). Cancer statistics, 2020. CA: A Cancer Journal for Clinicians, 70(1), 7-30, available: doi:10.3322/caac.21590
Spigel, D. R., & Socinski, M. A. (2013). Rationale for chemotherapy, immunotherapy, and checkpoint blockade in SCLC: Beyond traditional treatment approaches. Journal of Thoracic Oncology, 8(5), 587-598, available: doi:10.1097/jto.0b013e318286cf88
Stafford, W. C., Peng, X., Olofsson, M. H., Zhang, X., Luci, D. K., Lu, L., Cheng, Q., Trésaugues, L., Dexheimer, T. S., Coussens, N. P., Augsten, M., Ahlzén, H. M., Orwar, O., Ostam, A., Stone-Elander, S., Maloney, D. J., Jadhav, A., Simeonov, A., Linder, S., & Arnér, E. S. (2018). Irreversible inhibition of cytosolic thioredoxin reductase 1 as a mechanistic basis for anticancer therapy. Science Translational Medicine, 10(428), available: doi:10.1126/scitranslmed.aaf7444
Stewart, C. A., Gay, C. M., Xi, Y., Sivajothi, S., Sivakamasundari, V., Fujimoto, J., Bolisetty, M., Hartsfield, P. M., Balasubramaniyan, V., Chalishazar, M. D., Moran, C., Kalhor, N., Strewart, J., Tran, H., Swisher, S. G., Roth, J. A., Zhang, J., Groot, J., Glisson, B., Oliver, T. G., Heymach, J. V., Wistuba, I., Robson, P., Wang, J., & Byers, L. A. (2020). Single-cell analyses reveal increased intratumoral heterogeneity after the onset of therapy resistance in small-cell lung cancer. Nature Cancer, 1(4), 423-436, available: doi:10.1038/s43018-019-0020-z
Subramanian, A., Tamayo, P., Mootha, V. K., Mukherjee, S., Ebert, B. L., Gillette, M. A., Paulovich, A., Pomeroy, S. L., Golub, T. R., Lander, E. S., & Mesirov, J. P. (2005). Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proceedings of the National Academy of Sciences, 102(43), 15545-15550, available: doi:10.1073/pnas.0506580102
Tang, Z., Kang, B., Li, C., Chen, T., & Zhang, Z. (2019). GEPIA2: An enhanced web server for large-scale expression profiling and interactive analysis. Nucleic Acids Research, 47(W1), 556-560, available: doi:10.1093/nar/gkz430
Waqar, S. N., & Morgensztern, D. (2017). Treatment advances in small cell lung cancer (SCLC). Pharmacology & Therapeutics, 180, 16-23, available: doi:10.1016/j.pharmthera.2017.06.002
Weng, M., Chang, J., Hung, W., Yang, Y., & Chien, M. (2018). The interplay of reactive oxygen species and the epidermal growth factor receptor in tumor progression and drug resistance. Journal of Experimental & Clinical Cancer Research, 37(1), available: doi:10.1186/s13046-018-0728-0
Xu, M., Xu, H., Chen, J., Chen, C., Xu, F., & Qin, Z. (2018). Generation of conditional Acvrl1 knockout mice by CRISPR/Cas9-mediated gene targeting. Molecular and Cellular Probes, 37, 32-38, available: doi:10.1016/j.mcp.2017.11.003
Yokouchi, H., Nishihara, H., Harada, T., Yamazaki, S., Kikuchi, H., Oizumi, S., Uramoto, H., Tanaka, F., Harada, M., Akie, K., Sugaya, F., Fujita, Y., Takamura, K., Kojima, T., Higuchi, M., Honjo, O., Minami, Y., Watanbe, N., Nishimura, M., Suzukim H., Dosaka-Akita, H., & Isobe, H. (2020). Detection of somatic TP53 mutation in surgically resected small-cell lung cancer by targeted exome sequencing: Association with longer relapse-free survival. Heliyon, 6(7), available: doi:10.1016/j.heliyon.2020.e04439
Zhang, X., Lan, Y., Xu, J., Quan, F., Zhao, E., Deng, C., Luo, T., Xu, L., Liao, G., Yan, M., Ping, Y., Li, F., Shi, A., Bai, J., Zhao, T., Li, X., & Xiao, Y. (2018). CellMarker: A manually curated resource of cell markers in human and mouse. Nucleic Acids Research, 47(D1), available: doi:10.1093/nar/gky900
Zhou, J., Li, Y., Li, D., Liu, Z., & Zhang, J. (2019). Oncoprotein LAMTOR5 activates GLUT1 via upregulating NF-κB in liver cancer. Open Medicine, 14(1), 264-270, available: doi:10.1515/med-2019-0022
Zhu, B., Ren, C., Du, K., Zhu, H., Ai, Y., Kang, F., Luo, Y., Liu, W., Wang, L., Xu, Y., Jiang, X., & Zhang, Y. (2019). Olean-28,13b-olide 2 plays a role in cisplatin-mediated apoptosis and reverses cisplatin resistance in human lung cancer through multiple signaling pathways. Biochemical Pharmacology, 170, 113642, available: doi:10.1016/j.bcp.2019.113642