
Author: Jake E. Batchelder
Autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy (APECED) is a rare, recessive disease caused by mutations in the autoimmune regulator (AIRE) gene. A loss of function at the AIRE locus is widely known to induce autoimmune activation against host tissues due to lack of central tolerance during thymic T cell development. Failure to delete autoreactive T cell clones allows their release into the periphery, where they may proliferate and initiate an autoimmune response. While APECED is a monogenic disorder, disruption of AIRE function can have diverse implications: similar mutations in AIRE can lead to a myriad of phenotypes and symptoms. By investigating the multiple ways AIRE function can be compromised, recent research has uncovered the steadfast mechanisms explaining how AIRE is expressed in mTECs, how AIRE transactivates tissue-specific antigens (TSAs), and how those TSAs are presented to T cells by both medullary thymic epithelial cells (mTECs) and bone marrow-derived antigen-presenting cells. However, the stochastic nature of APECED symptoms remains. Therefore, new approaches to APECED therapy should investigate the intersection of pragmatism and randomness inherent in the relationship between central and peripheral tolerance.
Introduction
T cells provide capable, targeted defense against foreign antigens through their receptor specificity. The vast repertoire of T cell receptors allows the immune system to mount a response against most foreign invaders. Generation of receptor diversity is accomplished mainly through gene rearrangement at the alpha and beta chain loci.
Positive selection in the thymic cortex is able to expand T cell clones with receptors that bind major histocompatibility complex (MHC)/self-peptide complexes with at least moderate affinity (De Martino et al., 2013). However, cells that pass positive selection may still have a strong affinity for self-peptides presented on MHC molecules. In order to eliminate these autoreactive T cells from escaping from the thymus into the periphery, T cell clones positively selected for in the thymic cortex undergo negative selection in the thymic medulla. During the negative selection process, T cells are presented with medullary thymic epithelial cell (mTEC)-expressed tissue-specific antigens (TSAs) in the medulla (Derbinski, Schulte, Kyewski, & Klein, 2001; Kyewski & Derbinski, 2004). T cells that show strong affinity for these self-peptide/MHC complexes are deleted by activation-induced apoptosis. The deletion of autoreactive T cell clones through thymic-expressed TSAs is known as central tolerance.
The discrepancy between antigens expressed and presented by cortical thymic epithelial cells (cTECs) versus mTECs has been termed the alternate peptide hypothesis. This hypothesis can partially explain how autoreactive T cells survive positive selection in the cortex but fail to pass negative selection in the medulla (Marrack, McCormack, & Kappler, 1989). In order to express TSAs, mTECs must transactivate genes that are not normally expressed in the thymus through a process called promiscuous gene expression (PGE; De Martino et al., 2013; Kyewski & Derbinski, 2004; Laan & Peterson, 2013; Metzger & Anderson, 2011; Tykocinski, Sinemus, & Kyewski, 2008). PGE is dependent upon the transcription of DNA in chromatin states often associated with inhibited expression (Abramson, Giraud, Benoist, & Mathis, 2010; Tykocinski et al., 2010; Ucar & Rattay, 2015; Žumer, Saksela, & Peterlin, 2013). The autoimmune regulator (AIRE) protein expressed in mTECs is a transcription factor that facilitates this process.
Loss of AIRE function limits TSA tolerance, leading to organ-specific autoimmunity and autoantibody production (Kisand & Peterson, 2015; Laan & Peterson, 2013; Metzger & Anderson, 2011). Autoimmune polyendocrinopathy candidiasis ectodermal dystrophy (APECED) is the monogenic disorder caused by mutations at the AIRE locus. However, APECED may be considered a syndrome because symptoms can also stem from indirect disruptions of AIRE function (De Martino et al., 2013). While APECED cases may feature some similar symptoms such as mucocutaneous candidiasis, Addison’s disease, and hypoparathyroidism, AIRE’s role in maintaining central tolerance to most TSAs makes APECED patients susceptible to further autoimmune responses against a number of host tissues including the eyes, liver, pancreas, kidney, and sex organs (Kisand & Peterson, 2015; Kyewski & Derbinski, 2004). Therefore, even APECED patients with similar mutations at the AIRE locus may have dissimilar symptoms due to variation in specific self-antigen tolerance (De Martino et al., 2013).
Because disruption of AIRE function can occur in numerous ways, this review will discuss how disruptions in AIRE expression, PGE promotion, and TSA presentation can all instigate auto-immunity. Furthermore, this review will explain how the interplay between central and peripheral tolerance contributes to the variation seen in APECED phenotypes and symptoms.
mTEC Development, Epigenetic Profile, and miRNA Govern AIRE Expression
Expression of AIRE is vital to expression of self-antigens in the thymus. AIRE is predominantly expressed in mTECs, although other cell types in the periphery and thymus have been shown to express AIRE at low levels (Derbinski et al., 2001; Metzger & Anderson, 2011). While mTEC lineage cells are primarily responsible for PGE, only 1-5% of mTEC cells express TSAs at a given time (Gallegos & Bevan, 2004). It is likely that factors unlinked to AIRE expression levels, such as changes in signaling states within the medullary microenvironment, alter PGE in AIRE+ mTECs. Nonetheless, because AIRE plays a direct role in PGE, disruption of AIRE expression leads to autoimmune phenotypes.
Problems with AIRE expression in mTECs may arise from impediments to mature mTEC development. Immature mTECs begin at the MHC IIlow, CD80low, AIRE- stage and mature to an MHC II-high, CD80high, AIRE- stage and then an MHC IIhigh, CD80high, AIRE+ stage, at which point AIRE-dependent and AIRE-independent antigens can be expressed (Metzger & Anderson, 2011). While this three-step maturation process represents the development needed for mTECs to activate PGE, mTECs have recently been shown to lose their AIRE+ phenotype during a fourth, and final, maturation stage. This loss of AIRE expression is coupled with a loss of TSA expression in these mature mTECs (Laan & Peterson, 2013; Yano et al., 2008). Therefore, while the paucity of TSA expression by the total mTEC population likely hinges on many factors, one possible contributor may be the finite timeframe in which AIRE is expressed by maturing mTECs.
While AIRE propels mTEC maturation, disruptions to early mTEC development stunt AIRE expression. AIRE-deficient mice produce malformed thymi, which illustrates the important role AIRE plays in thymic formation and mTEC development (Yano et al., 2008). However, failure of immature mTECs to pass proper developmental stages can have a substantial impact on AIRE expression and PGE in the thymus. For example, Rossi et al. (2007) show that RANK signaling from CD4+, CD3- cells facilitates mTEC development and promotes AIRE+ phenotypes. Absence of RANK signaling was shown to incite autoimmunity (Rossi et al., 2007). Later experiments in vivo uncovered that RANK signaling regulates AIRE function by promoting its accumulation within chromatin-associated nuclear bodies (Ferguson et al., 2008).
Failure to express AIRE may also stem from improper epigenetic markers at the AIRE locus. One epigenetic marker that influences gene expression is methylation of DNA. Hypermethylation of DNA can occur in contiguous regions, such as CpG islands, and is associated with low expression rates. Bisulfate sequencing of CpG islands near the AIRE promoter revealed hypomethylation surrounding the AIRE promoter in AIRE+ mTECS. However, these hypomethylation markers were also found in AIRE- immature (MHC IIlow) mTECs and cTECs, illustrating that AIRE promoter methylation likely has little effect on AIRE expression (Kont et al., 2011; Ucar & Rattay, 2015).
Alterations in the packing and chemical modification of chromatin can also dictate levels of gene expression. DNA is packed into nucleosomes, which contain DNA looped around octamers of histone proteins, similar to beads on a string. Tightly packed chromatin (heterochromatin) can sterically hinder transcriptional machinery from accessing promoters and other DNA sequences, making heterochromatin states unreceptive to gene expression. Conversely, loosely packed chromatin (euchromatin) is permissive of transcriptional machinery and gene expression. Furthermore, methylation of lysine residues on individual histone proteins can also promote or repress transcription. Histone profiling at the AIRE promoter in AIRE+ mTECs showed increased amounts of transcriptionally active histone marks (H3K4me3) and lower amounts of repressive histone marks (H3K27me3) than other cell types (Kont et al., 2011). These epigenetic patterns illustrate that AIRE expression is correlated with histone modifications at the promoter region. Failure to properly mark specific histone residues at the AIRE promoter may cause epigenetic silencing of AIRE, leading to decreased PGE and autoimmune phenotypes.
Expression of AIRE may be further regulated by miRNA interactions. Research by Ucar, Tykocinski, Dooley, Liston, and Kyewski (2013). revealed that miRNAs are tightly regulated in developing mTECs. In addition, mice lacking Dicer function showed loss of AIRE expression and reduced PGE, which demonstrates that miRNA regulates AIRE expression and function (Ucar et al., 2013). Therefore, loss of AIRE function may stem from an inability of miRNA to regulate AIRE activity.
Disruptions of AIRE or Other Promiscuous Gene Expression Mediators Yield Autoimmunity
AIRE contributes to central tolerance by enabling the expression of self-antigens within mTECs through PGE. Promoting TSA expression is a complex process, and AIRE is able to facilitate PGE through its unique protein domains, which allow for subcellular localization and interaction with other proteins that assist in the transcription and processing of TSAs (Abramson et al., 2010; De Martino et al., 2013; Gallo et al., 2013; Ramsey, Bukrinsky, & Peltonen, 2002). Taken together, mutations in the AIRE locus compromise the function of AIRE protein domains and lead to nonfunctional PGE.
In order to facilitate the transcription of TSAs not canonically expressed in the thymus, AIRE must localize to genes that are epigenetically repressed. AIRE protein domains allow it to access repressive chromatin states and transactivate TSA expression. For example, a dominant missense mutation in the SAND domain inhibited PGE in heterozygous mice by impeding localization of AIRE proteins encoded by both alleles to nuclear bodies. This mutation was sufficient to prompt an autoimmune phenotype (Su et al., 2008). Mutations in the CARD domain limited AIRE homodimerization and nuclear localization in vitro (Ferguson et al., 2008; Metzger & Anderson, 2011). Mutations leading to elimination of the AIRE C-terminus barred TSA expression by preventing AIRE from interacting with positive transcription elongation factor B (P-TEFb; Žumer, Plemenitaš, Saksela, & Peterlin, 2011). Synthetic mutations in the PHD domain revealed that the BHC80 region of AIRE’s PHD1 domain is vital for localization to nucleosomes. The PHD1 domain is a protein-binding zinc finger, which can bind hypomethylated H3K4, a traditionally repressive histone mark, in order to allow transcription within regions of heterochromatin (Anderson & Su, 2016). While AIRE binding of hypomethylated histone H3 tails was necessary for PGE, overexpression of H3K4-demethylase did not increase PGE, indicating that AIRE’s targets other epigenetic modifications (Koh, Kingston, Benoist, & Mathis, 2010). This hypothesis was supported by Waterfield et al. (2014), who used a screening approach to demonstrate that AIRE interacts with MBD1on its SAND domain. MDB1 is able to bind methylated CpG dinucleotides, which allows AIRE to localize to genes located within hypermethylated CpG islands (Waterfield et al., 2014).
Subcellular localization of AIRE to epigenetically-repressed sites via its protein domains is necessary for AIRE to facilitate the transcription of TSA genes. However, further protein-protein interactions also contribute to TSA transactivation. Because of AIRE’s integral role in facilitating a process that breaks conventional guidelines of gene regulation, it may be assumed that AIRE acts as a “pioneer protein,” which recruits RNA Polymerase II to TSA loci amidst a jumble of heterochromatin and other repressive epigenetic marks. However, Giraud et al. (2012) showed that the absence of AIRE did not inhibit expression of the first exon in AIRE-targeted genes. This illustrates that AIRE is not necessary for RNA Polymerase II to access epigenetically-repressed loci. Instead, RNA Polymerase II can be recruited to these sites by DNA-Dependent Protein Kinase (DNA-PK) in response to double stranded breaks caused by Topoisomerase II activity. RNA Polymerase II is then able to begin transcription of the first exon, but elongation is halted by negative elongation factors. AIRE also interacts with DNA-PK, which allows it to co-localize with RNA Polymerase II. After co-localization, AIRE’s interaction with P-TEFb prompts RNA polymerase II phosphorylation and transcriptional elongation (Žumer et al., 2013). Therefore, instead of initiating transcription at TSA loci, AIRE works to promote TSA transcription by unleashing RNA Polymerase II in order to transcribe downstream exons (Giraud et al., 2012). AIRE localization to double stranded break repair sites via DNA-PK provides a viable explanation to how AIRE accesses epigenetically-repressed TSA loci. However, as illustrated above, mutations to multiple AIRE protein domains have also been shown to inhibit subcellular localization and provoke autoimmune phenotypes. Further research will need to define whether these various methods of TSA localization work in tandem or in isolation to induce PGE.
AIRE also regulates TSA output via post-translational mRNA splicing (Kyewski & Derbinski, 2004; Žumer et al., 2011). mTECs had the greatest amount of alternatively spliced isoforms compared to any other cell type (Keane, Ceredig, & Seoighe, 2015). AIRE is thought to recruit splicing machinery in multiple ways. For example, splice factor snRNP is known to localize to nuclear bodies, (Sleeman & Lamond, 1999) where AIRE is also recruited via its SAND domain (Ramsey et al., 2002). Furthermore, Zumer et al. (2011) showed that snRNP subunit U5 was recruited by AIRE to the 3’ end of TSA transcripts. Therefore, AIRE utilizes co-localization with RNA Polymerase II to promote mRNA splicing (Žumer et al., 2011). AIRE is thought to perform mRNA splicing in order to tolerize autoreactive T cells specific to particular TSA isoforms, thus increasing the breadth of clonal deletion in the thymus (Keane et al., 2015; Kyewski & Derbinski, 2004). Therefore, disruption of mRNA splicing mechanics may limit the breadth of PGE expression, leading to autoimmunity of specific self-peptide isoforms.
mTECs and Thymic Dendritic Cells Present Tissue-Specific Antigens to Induce Tolerance
While TSA expression in mTECs is necessary for negative selection, central tolerance can be accomplished only if those TSAs are presented to T cells via MHC molecules. Therefore, failure to regulate TSA presentation on thymic cell types may undermine AIRE function and cause autoimmune phenotypes. Although mTECs have the proper surface molecules to initiate activation-induced apoptosis in both CD4+ and CD8+ T cells (Laan & Peterson, 2013), mTECs share these presenting responsibilities with thymic dendritic cells. These dendritic cells can be recruited to the thymus through the XC-chemokine ligand 1 (XCL1), a protein that is expressed by AIRE+ mTECs (Anderson & Su, 2016). It should be noted that thymic dendritic cells do not express AIRE and do not perform PGE (Derbinski et al., 2001). Therefore, mTECs serve as TSA reservoirs, and can selectively pass off PGE products to thymic dendritic cells for presentation (Gallegos & Bevan, 2004; Hubert et al., 2011; Metzger & Anderson, 2011).
By regulating thymic and bone marrow expression of ovalbumin (OVA) peptide and MHC I/II, respectively, in mouse models, researchers have investigated whether mTECs are self-sufficient at inducing autoreactive CD4+ and/or CD8+ T cell deletion through TSA presentation. TSAs produced by mTECs are intracellular proteins, and should therefore be canonically presented by MHC I to CD8+ T cells; mTEC presentation to CD4+ T cells would require cross-presentation of intracellular TSAs to MHC II. Gallegos and Bevan hypothesized that because mTECs were insufficient at antigen presentation, thymic dendritic cells were responsible for presentation to CD4+ and CD8+ T cells. Their results indicated that mTECs self-sufficiently induced CD8+ T cell tolerance to mOVA, but bone-marrow derived thymic dendritic cells were necessary for tolerance of mOVA-specific CD4+ T cells (Gallegos & Bevan, 2004). However, recent evidence has qualified those findings, asserting that mTECs are responsible for some TSA antigen via MHC II, but induction of CD4+ OVA tolerance is greatly diminished in mice with MHC II-deficient bone marrow (Hubert et al., 2011).
While inducing thymic expression of OVA through knock-in experiments spotlights the presentation responsibilities between mTECs and thymic dendritic cells for one non-self peptide, how presentation of numerous, specific TSAs is delineated between mTECs and thymic dendritic cells for comprehensive tolerance induction remains unknown. Research by Zhang et al. (2003) showed that soluble hen egg lysozyme(HEL) expression in the thymus produced more efficient negative selection of CD4+ thymocytes than membrane-bound HEL, suggesting mTEC secretion of peptides to thymic dendritic cells is important for tolerance induction. However, because autoreactive T cells are prone to interact with membrane-bound molecules on the surface of tissues, uncovering how shared presentation responsibilities ensure full tolerance to all self-peptides is an important step to uncovering more about negative selection mechanics.
Peripheral Tolerance Drives Variability in APECED Symptomatology
The disparities found in APECED symptomatology stem from the limited power of peripheral tolerance. Mechanisms of peripheral tolerance inactivate autoreactive lymphocytes that have escaped central tolerance during T lymphocyte development. For example, immature dendritic cells in the periphery are responsible for induction of tolerance to self-antigens under steady-state conditions (Hawiger et al., 2001; Mueller, 2010). Dendritic cells in both lymph nodes and the spleen can process, load, and present self-antigens from the periphery to T cells. Thus, expression of certain antigens in the periphery is sufficient to induce tolerance of those antigens (Derbinski et al., 2001). Additionally, certain dendritic cells express limited amounts of AIRE. These extrathymic AIRE-expressing cells (eTACs) may provide additional tolerance in basal conditions by presenting AIRE-dependent self-antigens in the periphery (Metzger & Anderson, 2011; Mueller, 2010). eTACs lack costimulatory molecules CD80/86, which may induce anergy in T cells that recognize eTAC-presented peptides (Metzger & Anderson, 2011). However, it is likely that eTAC levels are minimal in APECED patients.
Peripheral tolerance is also formed by regulatory T cells (CD4+, FOXP3+, CD25+), which induce anergy to helper and cytotoxic T cells through direct interaction, releasing anti-inflammatory signals, and expending cytokines that potentiate T cell activation and proliferation. T cells in the thymus may be pushed to the thymic regulatory T cell lineage if they bind MHC/self-peptide complexes with strong affinity during negative selection in the thymus (Jordan et al., 2001). Induced regulatory T cells may be induced to undergo lineage commitment in the periphery through receptor activation and epigenetic change at the FOXP3 locus (Ohkura et al., 2012). Because AIRE deficiency hinders negative selection through dysfunctional PGE, APECED patients possess limited regulatory T cell populations, likely due to the inability to facilitate thymic regulatory T cell lineage commitment (Kekäläinen et al., 2007; Perry et al., 2014). However, induced regulatory T cells may play a role in muffling the autoimmune response in some tissues.
The role of peripheral tolerance mechanisms to silence auto-immunity in host tissues causes the variability of APECED phenotypes. The stochastic nature of T cell receptor gene rearrangement leads to a diverse potential of peripheral autoreactive T cells in AIRE-deficient individuals (Kisand & Peterson, 2015). Peripheral tolerance serves to filter out those autoreactive T cell responses, but because the peripheral filter is imperfect, the list of specific autoreactive T cell clones left unconstrained is unpredictable (Figure 1). For example, the self-peptides available for dendritic cells to uptake and present may depend on random circumstance, leaving the peripheral tolerance of specific tissues up to chance. Furthermore, variability in the activation and recruitment of specific induced regulatory T cell clones further confounds which autoreactive helper and cytotoxic T cells will cause host tissue damage. Still other factors, such as the amount of costimulatory molecules and activation-inducing cytokines present in a given tissue, play further roles (Klein & Kyewski, 2000). Therefore, while dysfunctional PGE in the thymus is sufficient to promote a myriad of autoreactive T cells in the periphery of APECED patients, the variable phenotypes associated with the disorder result from the stochastic mechanisms of peripheral tolerance used to neutralize autoreactive activity.
Conclusions and Future Directions
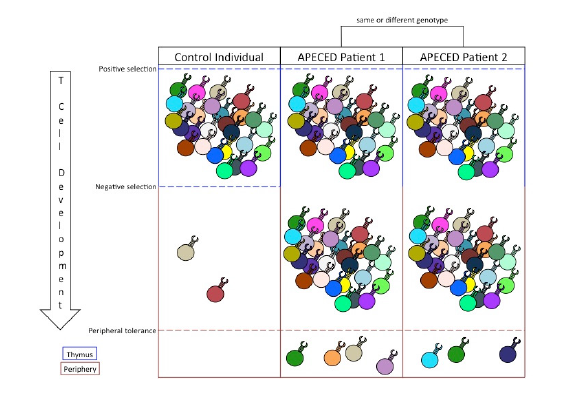
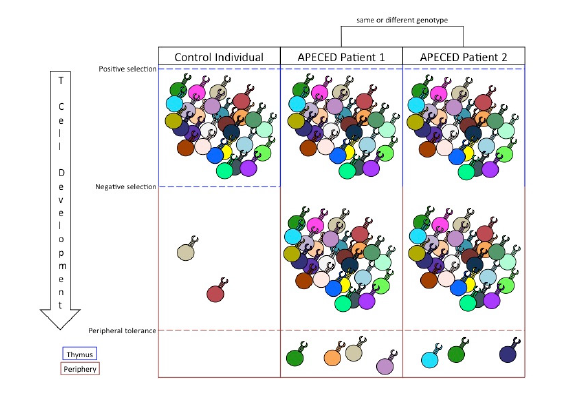
Figure 1. The relationship between central and peripheral tolerance determines the profile of autoreactive T cells in the periphery. Positive selection expands T cell clones that garner a signal from MHC/self-peptide complexes. Negative selection filters out autoreactive T cell clones that bind with high affinity to MHC/tissue-specific antigen complexes. Inability to perform negative selection permits autoreactive T cell clones into the periphery. Peripheral tolerance suppresses a limited number of autoreactive T cell responses and, in cases of APECED, dictates the specific autoimmune symptoms of the patient.
Because of AIRE’s central role in facilitating PGE, lack of central tolerance is intrinsic in every APECED phenotype (Figure 1). However, current treatment options do not remedy issues with central tolerance. Instead, treatments of APECED focus on maintaining tissue function and suppressing immune system responses through anti-inflammatory drugs. These treatments are often ineffective in limiting the autoimmune responses (Kisand & Peterson, 2015). While successful constitution of central tolerance in APECED patients would cure their symptoms, the complexity of the mechanisms involved in AIRE expression, PGE, and TSA presentation poses significant obstacles to targeting central tolerance therapeutically. Therefore, treatments for APECED patients could instead utilize the suppressive mechanisms of peripheral tolerance.
Peripheral tolerance is an effective suppressor of autoimmune responses. Despite the diversity of autoreactive T cells in the periphery of APECED patients, typical patients experience autoimmune responses to only a limited number of tissues (Figure 1; Kisand & Peterson, 2015). This is because peripheral tolerance is responsible for suppressing the activation, proliferation, and activity of autoreactive T cells. As such, an autoimmune response to any self-antigen can be thought of as a failure of peripheral tolerance to protect that antigen from immune targeting.
Identifying the autoimmune responses in each APECED patient inherently identifies the limits of peripheral tolerance in that individual. Therefore, new therapeutic efforts for APECED could address breaches in peripheral tolerance in a symptom-specific manner: patients would receive therapy that would induce peripheral tolerance to the tissues under attack. This treatment might be accomplished by introducing the self-antigens of interest to secondary lymphoid organs, where immature dendritic cells may tolerize peripheral T cells specific to those antigens; a 2012 study showed that mice injected with microparticles decorated with a specific antigen induced long term tolerance of T cells specific to that antigen (Getts et al., 2012). Additionally, transplanting tissue-specific regulatory T cells into the periphery may promote anergy to a given tissue. Many regulatory T cell-based therapeutic studies are currently in clinical trials, and future studies may utilize specific MHC/peptide combinations to isolate and expand antigen-specific regulatory T cells (Khor, 2016). While these methods of inducing tissue-specific peripheral tolerance are far from developed, they provide the potential to overcome the variability associated with both the causes and symptoms of APECED.
References
Abramson, J., Giraud, M., Benoist, C., & Mathis, D. (2010). Aire’s Partners in the Molecular Control of Immunological Tolerance Cell, 140(1), 123–135. doi:10.1016/j.cell.2009.12.030
Anderson, M. S., & Su, M. A. (2016). AIRE expands: new roles in immune tolerance
and beyond. Nature Reviews Immunology, 16(4), 247–258. doi:10.1038/
nri.2016.9
De Martino, L., Capalbo, D., Improda, N., D’Elia, F., Di Mase, R., D’Assante,
R., . . . Salerno, M. (2013). APECED: A Paradigm of Complex Interactions
between Genetic Background and Susceptibility Factors. Frontiers in Immunology,
4. doi:10.3389/fimmu.2013.00331
Derbinski, J., Schulte, A., Kyewski, B., & Klein, L. (2001). Promiscuous gene
expression in medullary thymic epithelial cells mirrors the peripheral self.
Nature Immunology, 2(11), 1032.
Ferguson, B. J., Alexander, C., Rossi, S. W., Liiv, I., Rebane, A., Worth, C. L., . .
.Rich, T. (2008). AIRE’s CARD Revealed, a New Structure for Central Tolerance
Provokes Transcriptional Plasticity. Journal of Biological Chemistry,
283(3), 1723–1731. doi:10.1074/jbc.M707211200
Gallegos, A. M., & Bevan, M. J. (2004). Central Tolerance to Tissue-specific Antigens
Mediated by Direct and Indirect Antigen Presentation. The Journal
of Experimental Medicine, 200(8), 1039–1049. doi:10.1084/jem.20041457
Gallo, V., Giardino, G., Capalbo, D., Palamaro, L., Romano, R., Santamaria, F., .
. . Pignata, C. (2013). Alterations of the autoimmune regulator transcription
factor and failure of central tolerance: APECED as a model. Expert Review
of Clinical Immunology, 9(1), 43–51. doi:10.1586/eci.12.88
Getts, D. R., Martin, A. J., McCarthy, D. P., Terry, R. L., Hunter, Z. N., Yap, W. T.,
. . . Miller, S. D. (2012). Microparticles bearing encephalitogenic peptides
induce T-cell tolerance and ameliorate experimental autoimmune encephalomyelitis.
Nature Biotechnology, 30(12), 1217–1224. doi:10.1038/nbt.2434
Giraud, M., Yoshida, H., Abramson, J., Rahl, P. B., Young, R. A., Mathis, D., &
Benoist, C. (2012). Aire unleashes stalled RNA polymerase to induce ectopic
gene expression in thymic epithelial cells. Proceedings of the National Academy
of Sciences, 109(2), 535–540. doi:10.1073/pnas.1119351109
Hawiger, D., Inaba, K., Dorsett, Y., Guo, M., Mahnke, K., Rivera, M., . . . Nussenzweig,
M. C. (2001). Dendritic Cells Induce Peripheral T Cell Unresponsiveness
under Steady State Conditions in Vivo. The Journal of Experimental
Medicine, 194(6), 769–780. doi:10.1084/jem.194.6.769
Hubert, F.-X., Kinkel, S. A., Davey, G. M., Phipson, B., Mueller, S. N., Liston, A., .
. . Heath, W. R. (2011). Aire regulates the transfer of antigen from mTECs to
dendritic cells for induction of thymic tolerance. Blood, 118(9), 2462–2472.
doi:10.1182/blood-2010-06-286393
Jordan, M. S., Boesteanu, A., Reed, A. J., Petrone, A. L., Holenbeck, A. E., Lerman,
M. A., . . . Caton, A. J. (2001). Thymic selection of CD4+CD25+ regulatory
T cells induced by an agonist self-peptide. Nature Immunology, 2(4), 301.
Keane, P., Ceredig, R., & Seoighe, C. (2015). Promiscuous mRNA splicing under
the control of AIRE in medullary thymic epithelial cells. Bioinformatics,
31(7), 986–990. doi:10.1093/bioinformatics/btu785
Kekäläinen, E., Tuovinen, H., Joensuu, J., Gylling, M., Franssila, R., Pöntynen, N.,
. . . Arstila, T. P. (2007). A Defect of Regulatory T Cells in Patients with Autoimmune
Polyendocrinopathy-Candidiasis-Ectodermal Dystrophy. The Journal
of Immunology, 178(2), 1208–1215. doi:10.4049/jimmunol.178.2.1208
Khor, B. (2016). Regulatory T cells: Central concepts from ontogeny to therapy.
Transfusion Medicine Reviews, 0(0). doi:10.1016/j.tmrv.2016.07.003
Kisand, K., & Peterson, P. (2015). Autoimmune Polyendocrinopathy Candidiasis
Ectodermal Dystrophy. Journal of Clinical Immunology, 35(5), 463–478.
doi:10.1007/s10875-015-0176-y
Klein, L., & Kyewski, B. (2000). Promiscuous expression of tissue antigens in the
thymus: a key to T-cell tolerance and autoimmunity? Journal of Molecular
Medicine, 78(9), 483–494.
Koh, A. S., Kingston, R. E., Benoist, C., & Mathis, D. (2010). Global relevance
of Aire binding to hypomethylated lysine-4 of histone-3. Proceedings of
the National Academy of Sciences, 107(29), 13016–13021. doi:10.1073/
pnas.1004436107
Kont, V., Murumägi, A., Tykocinski, L.-O., Kinkel, S. A., Webster, K. E., Kisand,
K., . . . Peterson, P. (2011). DNA methylation signatures of the AIRE promoter
in thymic epithelial cells, thymomas and normal tissues. Molecular
Immunology, 49(3), 518–526.
Kyewski, B., & Derbinski, J. (2004). Self-representation in the thymus: an extended
view. Nature Reviews Immunology, 4(9), 688–698. doi:10.1038/nri1436
Laan, M., & Peterson, P. (2013). The Many Faces of Aire in Central Tolerance.
Frontiers in Immunology, 4. doi:10.3389/fimmu.2013.00326
Marrack, P., McCormack, J., & Kappler, J. (1989). Presentation of antigen, foreign
major histocompatibility complex proteins and self by thymus cortical epithelium.
Nature, 338(6215), 503–505. doi:10.1038/338503a0
Metzger, T. C., & Anderson, M. S. (2011). Control of central and peripheral tolerance
by Aire. Immunological Reviews, 241(1), 89–103. doi:10.1111/j.1600-
065X.2011.01008.x
Mueller, D. L. (2010). Mechanisms maintaining peripheral tolerance. Nature Immunology,
11(1), 21–27. doi:10.1038/ni.1817
Ohkura, N., Hamaguchi, M., Morikawa, H., Sugimura, K., Tanaka, A., Ito, Y., .
. . Sakaguchi, S. (2012). T Cell Receptor Stimulation-Induced Epigenetic
Changes and Foxp3 Expression Are Independent and Complementary
Events Required for Treg Cell Development. Immunity, 37(5), 785–799.
doi:10.1016/j.immuni.2012.09.010
Perry, J. S. A., Lio, C.-W. J., Kau, A. L., Nutsch, K., Yang, Z., Gordon, J. I., . . .
Hsieh, C.-S. (2014). Distinct contributions of Aire and antigen presenting cell
subsets to the generation of self-tolerance in the thymus. Immunity, 41(3),
414–426. doi:10.1016/j.immuni.2014.08.007
Ramsey, C., Bukrinsky, A., & Peltonen, L. (2002). Systematic mutagenesis of the
functional domains of AIRE reveals their role in intracellular targeting. Human
Molecular Genetics, 11(26), 3299–3308. doi:10.1093/hmg/11.26.3299
Rossi, S. W., Kim, M.-Y., Leibbrandt, A., Parnell, S. M., Jenkinson, W. E., Glanville,
S. H., . . . Anderson, G. (2007). RANK signals from CD4+3− inducer
cells regulate development of Aire-expressing epithelial cells in the thymic
medulla. The Journal of Experimental Medicine, 204(6), 1267–1272.
doi:10.1084/jem.20062497
Sleeman, J. E., & Lamond, A. I. (1999). Newly assembled snRNPs associate
with coiled bodies before speckles, suggesting a nuclear snRNP maturation
pathway. Current Biology, 9(19), 1065–1074. doi:10.1016/S0960-
9822(99)80475-8
Su, M. A., Giang, K., Žumer, K., Jiang, H., Oven, I., Rinn, J. L., . . . Anderson,
M. S. (2008). Mechanisms of an autoimmunity syndrome in mice caused by
a dominant mutation in Aire. The Journal of Clinical Investigation, 118(5),
1712–1726. doi:10.1172/JCI34523
Tykocinski, L.-O., Sinemus, A., & Kyewski, B. (2008). The Thymus Medulla
Slowly Yields Its Secrets. Annals of the New York Academy of Sciences,
1143(1), 105–122. doi:10.1196/annals.1443.018
Tykocinski, L.-O., Sinemus, A., Rezavandy, E., Weiland, Y., Baddeley, D., Cremer,
C., . . . Kyewski, B. (2010). Epigenetic regulation of promiscuous gene
expression in thymic medullary epithelial cells. Proceedings of the National
Academy of Sciences, 107(45), 19426–19431. doi:10.1073/pnas.1009265107
Ucar, O., & Rattay, K. (2015). Promiscuous Gene Expression in the Thymus:
A Matter of Epigenetics, miRNA, and More? Frontiers in Immunology, 6.
doi:10.3389/fimmu.2015.00093
Ucar, O., Tykocinski, L.-O., Dooley, J., Liston, A., & Kyewski, B. (2013). An evolutionarily
conserved mutual interdependence between Aire and microRNAs
in promiscuous gene expression. European Journal of Immunology, 43(7),1769–1778. doi:10.1002/eji.201343343
Waterfield, M., Khan, I. S., Cortez, J. T., Fan, U., Metzger, T., Greer, A., . . . Anderson,
M. S. (2014). The transcriptional regulator Aire coopts the repressive
ATF7ip-MBD1 complex for the induction of immunotolerance. Nature Immunology,
15(3), 258–265. doi:10.1038/ni.2820
Yano, M., Kuroda, N., Han, H., Meguro-Horike, M., Nishikawa, Y., Kiyonari, H.,
. . . Matsumoto, M. (2008). Aire controls the differentiation program of thymic
epithelial cells in the medulla for the establishment of self-tolerance.
The Journal of Experimental Medicine, 205(12), 2827–2838. doi:10.1084/
jem.20080046
Zhang, M., Vacchio, M. S., Vistica, B. P., Lesage, S., Egwuagu, C. E., Yu, C.-R., . . .
Gery, I. (2003). T Cell Tolerance to a Neo-Self Antigen Expressed by Thymic
Epithelial Cells: The Soluble Form is More Effective Than the Membrane-
Bound Form. The Journal of Immunology, 170(8), 3954-3962. doi:10.4049/
jimmunol.170.8.3954
Žumer, K., Plemenitaš, A., Saksela, K., & Peterlin, B. M. (2011). Patient mutation
in AIRE disrupts P-TEFb binding and target gene transcription. Nucleic Acids
Research, 39(18), 7908–7919. doi:10.1093/nar/gkr527
Žumer, K., Saksela, K., & Peterlin, B. M. (2013). The Mechanism of Tissue-
Restricted Antigen Gene Expression by AIRE. The Journal of Immunology,
190(6), 2479–2482. doi:10.4049/jimmunol.1203210