
Authors: Cecily Wing Hei Cheng, Matthew Wai Heng Chung, & Joseph Chi Fung Ng
Institution: [1] School of Biomedical Sciences, Li Ka Shing Faculty of Medicine, University of Hong Kong, 3/F, Laboratory Block, 21 Sassoon Road, Pokfulam, Hong Kong; [2] Randall Division of Cell and Molecular Biophysics, King’s College, Room 3.14, New Hunt’s House, Guy’s Campus, London, SE1 1UL, United Kingdom.
Date: December 2016
doi: 10.22186/jyi.31.6.44-50
ABSTRACT
The nucleation of amyloid-β (Aβ) oligomers, and the fibril formation that follows represents an important pathologic mechanism for Alzheimer's disease (AD). This has motivated the search for therapeutics that specifically target Aβ, which holds promise to be a cure for AD. However, conventional biophysical approaches like X-ray crystallography and nuclear magnetic resonance (NMR) spectroscopy fall short of capturing this highly dynamic process. The aggregation of amyloid fibrils has been unravelled by a mix of novel approaches. For example, computational methods like molecular dynamics (MD) simulations provide atomistic predictions of structural dynamics of protein fibrils. On the other hand, various spectroscopy and spectrometry approaches allow unprecedented resolution and details to be experimentally observed and complement predictions offered by computational investigations. This review surveys these approaches in the context of studying Aβ peptide aggregation. We shall discuss how these methods help us understand the mechanistic aspect of the aggregative process. We emphasise that computational and experimental approaches must go together to obtain a comprehensive view on the structural dynamics of critical protein players in health and diseases.
INTRODUCTION
Aβ aggregates as causative agent for Alzheimer’s disease
AD is the most common form of dementia, featuring irreversible memory loss and deterioration in linguistic, cognitive, and learning capacities. Insoluble Aβ plaques are a hallmark of AD, with elevated levels of the 42-residue Aβ42 in the brain compared to a more abundant Aβ40 in healthy individuals (Näslund et al., 1994). Researchers have long followed the path of “amyloid hypothesis” in finding AD’s pathologic mechanism. This hypothesis holds that aggregation and deposition of Aβ peptides are the earliest initiating factors of AD, leading to the formation of paired helical filaments of tau aggregates and eventually neuronal cell death (Karran, Mercken & De Strooper, 2011).
Both soluble Aβ oligomers and insoluble Aβ fibrils are neurotoxic. The former is a better correlate of cognitive dysfunction; Aβ oligomers block long-term potentiation in hippocampal neurons (Tomic, Pensalfini, Head & Glabe, 2009). In Aβ fibrils, β-strands of individual Aβ peptides aggregate to form cross-β structures with inter-strand hydrogen bonds (Figure 1A). This provides surfaces onto which Aβ peptides are favourably “docked-and-locked” (Karran et al., 2011). Therefore, it is intuitive for therapeutics to attempt preventing Aβ aggregates, in both oligomer and fibril states.
To design these therapeutic interventions, we need to thoroughly understand the nucleating step, stability, and reversibility of these aggregates. While techniques like NMR contributed greatly to studying the structure of Aβ fibrils, Aβ oligomer structure is less clear due to its heterogeneity, instability, and variability across experimental conditions (Cerasoli, Ryadnov & Austen, 2015) (Figure 1B).
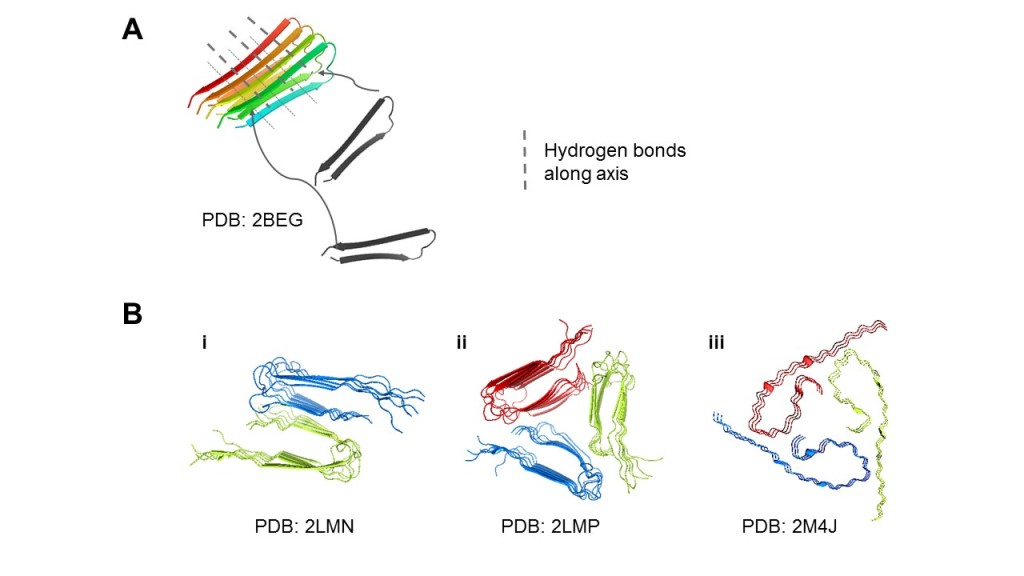
Figure 1. Aβ can aggregate into diverse structural polymorphs. (A) Microscopically, Aβ peptides arrange into β-structures by hydrogen bonds, and the aggregates form surfaces for more Aβ peptides to attach. (B) Structural polymorphs of Aβ40 solved by solid-state NMR. (i) 2-fold striated-ribbon Aβ9-40 prepared in vitro. (ii) 3-fold twisted Aβ9-40 prepared in vitro. (iii) AD patient-derived Aβ1-40 fibrils taken from brain tissue. We display different possible arrangements of the cross-β structures and flexible tails (i-iii), and the common 2- to 3-fold symmetry (i-ii). The accession codes for the relevant Protein Data Bank (PDB) structures used were noted.
The dynamic process of how Aβ is transiently misfolded, aggregates into oligomers, and subsequently forms protofibrils and fibrils is still elusive, due to the limitations of conventional biophysical techniques. In this review, we compare newer spectrometry and spectroscopy techniques with conventional biophysical approaches used in studying Aβ aggregation. In addition, computational strategies like MD simulation have generated insights into the dynamic properties of Aβ aggregates. We discuss their principles and usages and outline how a combination of experimental and computational methods contributes profound insights into Aβ aggregation. Both approaches should go hand in hand in order to understand, not only Aβ but critical peptides and proteins in human diseases.
Conventional Biophysical Techniques Only Give Snapshots of Aggregation
X-ray crystallography, NMR, and cryo-electron microscopy are established experimental methods to study protein structures and interaction at atomic resolution by capturing a particular instant of conformation. While they suffice to infer spatial arrangement and interactions, the study of time-dependent processes such as Aβ aggregation, which goes on for days, takes more than a few structural snapshots.
In the case of Aβ aggregation, solving a definitive structure is already a challenge. In its fibril state, Aβ becomes insoluble, and it is impossible to perform solution NMR or to grow crystals for typical X-ray diffraction analysis. It can at most be subjected to X-ray fibre diffraction at moderate resolution or solid-state NMR (ssNMR). Nevertheless, several sizes of Aβ oligomers can be soluble in sodium dodecyl sulphate (SDS), and such a sample had been subjected to solution NMR (Yu et al., 2009). These forms exhibit strong toxicity and neuropathogenicity (Ahmed et al., 2010). However, such solubilisation could shift the stabilization environment, possibly by adding detergent that does not properly reflect physiological situations. Moreover, the immense scale of Aβ aggregation complicates such inference (Robustelli, Stafford & Palmer, 2012).
Electron microscopy (EM) is one of the earliest approaches to studying amyloid fibrils (Shirahama & Cohen, 1967). Although EM has yielded insights into the basic structural features and might also capture several states of the aggregates, it is not meant to provide time-dependent data for analysis. In Aβ aggregation where the process is of utmost interest, EM, like NMR, is inadequate in understanding the dynamic details of Aβ fibril formation.
As shown in Figure 1B, amyloid fibrils can take on different morphology depending on the source. For example, in vitro-prepared fibrils can occur in varying layers. Although only one of the dozen conformations is displayed here, the remaining frames are merely a transient oscillation of flexible regions that highlight no key interaction events. Conventional methods thus leave us ignorant of processes that happened before and during aggregation, fragmentation, and secondary nucleation. As we are gradually convinced that Aβ aggregates can exist in any form, solving structures of each polymorph becomes less relevant and the spotlight shines on understanding and predicting how they are formed with molecular and biophysical details.
Molecular Dynamics Simulation can Computationally Model Aβ Aggregation
To overcome limitations of conventional biophysical techniques, computational MD simulations, provide a “time-lapse” dimension to the Aβ structure. Here we put the protein(s) of interest into a system surrounded with solvents and compute its/their movement by using Newton’s laws and sets of pre-defined parameters known as “force-fields”. These force-fields dictate the energy functions that model potential energies within bonded and non-bonded entities (Jernigan & Bahar, 1996). An all-atom conventional MD simulation of Aβ shows a trajectory from which inference can be drawn with regards to its stability and/or plasticity. Trajectories of wild-type and mutant protein can also be compared to identify critical residues in structure and function (Figure 2A). In the context of Aβ, a special variant of MD simulation known as steered MD (SMD) is particularly relevant. This is illustrated in Figure 2B, where the amyloid aggregate is taken as a control from which the fibril is pulled away. An “umbrella sampling” method samples conformations from varying distances of the pull to probe interaction across time (Lemkul & Bevan, 2010). This provides a detailed atomistic account of the reverse of aggregation, which is valuable in deciphering aggregation itself. Such framework can be enhanced to answer more specific biological questions. For example, robust free energy calculations performed on the SMD profile can quantify binding and aggregation of peptides (Perez, Morrone, Simmerling & Dill, 2016) (Figure 2B).
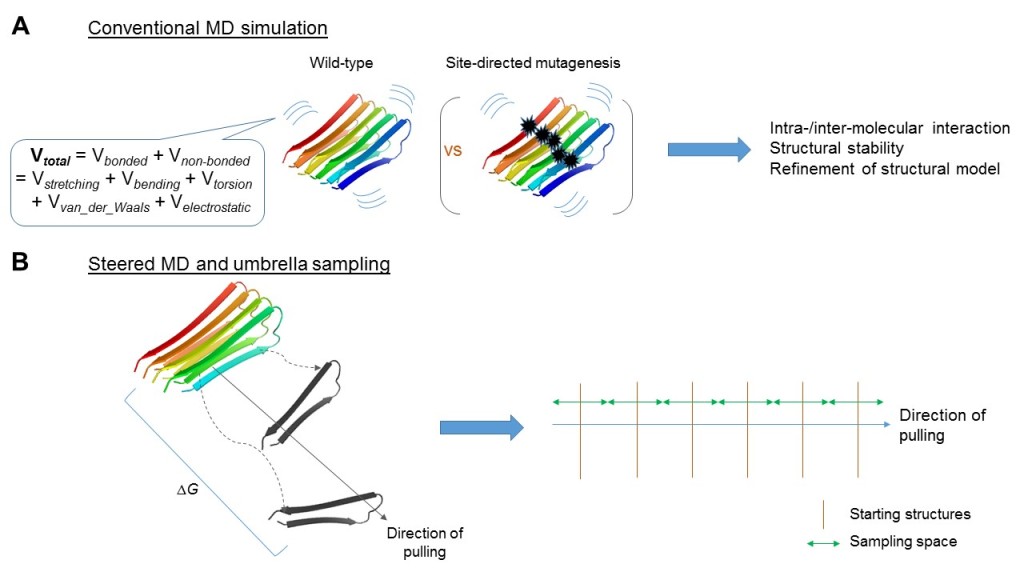
Figure 2. Conventional and steered molecular dynamics (MD) simulations address different questions. (A) Conventional MD simulation involves calculation of energy, both bonded and non-bonded, under parameterization defined by force-fields as exemplified. Coupling with, for example, site-directed mutagenesis, it is geared to study biochemical properties of the aggregates along the time-scale at atomistic resolution. (B) Alternatively, steered MD simulations, coupled with umbrella sampling could be used to address the aggregative process, e.g., by considering free energy changes (ΔG) throughout the process. PDB entry 2BEG was illustrated here.
An early paper manipulated simulations to characterise the conformational heterogeneity of a short stretch Aβ16-22 mediated by electrostatic and hydrophobic interactions (Klimov & Thirumalai, 2003). Lemkul and Bevan (2010) revealed a critical role for the salt bridge between Asp23 and Lys28 of Aβ42. They observed that this interaction was maintained throughout trajectories of wild-type amyloid fibres, and free energy calculations confirmed their role in stabilizing amyloid aggregates. While they accounted for intrinsic molecular details behind plasticity, Aβ aggregation was not directly addressed.
Kahler, Sticht and Horn (2013) have contributed more in this area. Their simulations revealed that Aβ fibrils in larger aggregates twisted at larger angles, and large aggregates subsequently broke. Smaller fragments then emerged as seeds to propagate aggregation. Recent research revealed that water plays a critical role in driving fibril assembly in Aβ40-seeded growth (Schwierz, Frost, Geissler, & Zacharias, 2016). These studies offered many insights, but their reliability has always been a matter of debate. As we shall see later, intrinsic changes of the surroundings seem to play a role in Aβ aggregation, especially concerning water in Aβ fibrils. It remains hard to factor in delicate contexts into the simulation, and this is where experimental approaches come in to validate such bioinformatics insights.
Spectroscopic and Spectrometric Techniques can Manipulate Real-Time Aβ Aggregation
A variety of spectroscopic/spectrometric techniques have emerged for investigating Aβ and other amyloid aggregation, offering high-resolution details of transient intermediates. Firstly, infrared spectroscopy relies on the excited vibrations of atoms in a molecule upon infrared radiation absorption (Figure 3A). While it is highly sensitive for antiparallel β-sheets, which are abundant in amyloids (Bruker Optics Inc, 2013), isotope labelling is needed to comprehend its complex spectrum. Usually, one or two carbonyl residues are labelled at a time by 13C and 18O (Arnaud, 2009), and the shifts of their peaks due to motions of neighbouring charges are also monitored. Another useful vibrational spectroscopy method is Raman spectroscopy (Figure 3A), which excites molecules to a higher virtual energy state and measures inelastic scattering of light (Kurouski, Van Duyne & Lednev, 2015). These spectroscopies have provided valuable information on the role of water and the disruption of lipid membranes. 2D infrared spectroscopy identified water neighbouring the amide groups of the hydrophobic Leu17 and Leu34 (Kim, Liu, Axelsen & Hochstrasser, 2009), which warranted more kinetic experiments to study its role. Raman spectroscopy showed that Aβ40 peptide within anionic lipid bilayers changes from disordered or helical conformations to β-sheets over time (Kurouski et al., 2015). However, since they can only collect averaged signals from the entire population of Aβ monomers, it cannot characterize individual conformations in the heterogeneous population at equilibrium as in mass spectrometry.
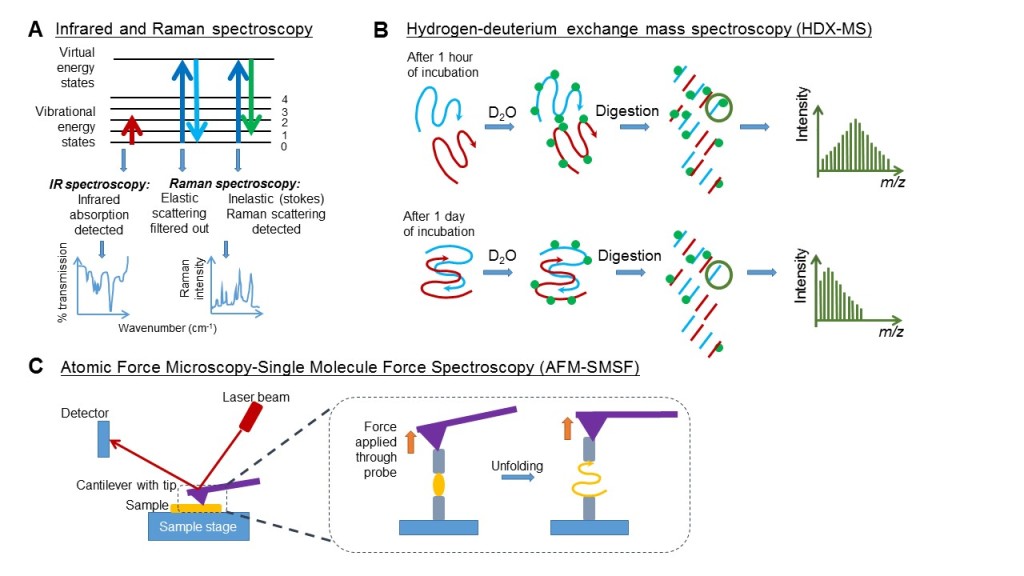
Figure 3. Different spectroscopy and spectrometry techniques vary in the principles employed to capture intrinsic changes within molecules. (A) Both Infrared and Raman spectroscopy involve excitation of the molecule to a higher energy state. The former detects the absorption of infrared radiation when the molecule is excited to a higher vibrational energy state. The latter detects the inelastic scattering of electromagnetic radiation when the molecule returns from a higher virtual energy state while retaining some photon energy. This inelastic scattering is a very rare occurrence detected only through filtering out the elastic scatterings simultaneously. (B) For HDX-MS, when the peptides (blue and red) are still monomers, HDX (deuterium as green dots) occurs rapidly along the peptide backbone. As incubation progresses, peptides start to aggregate, and the core is protected from rapid HDX. MS analysis at these two time-points numerates differences. m/z, mass-charge ratio. (C) In AFM-SMSF, the cantilever beam tip serves as a probe to gauge the topography and to apply force to measure mechanical stability/elasticity of peptide aggregates.
Figure 3B shows a particularly relevant mass spectrometry for Aβ aggregation dynamics based on hydrogen-deuterium exchange (HDX-MS), in which backbone amide hydrogens, which are exposed to solvent comprising “heavy water” (deuterium oxide) as a result of protein unfolding or hydrogen-bond-breaking, are replaced by deuterium (Eyles & Kaltashov, 2004). Each protium-deuterium exchange will increase the protein mass by a single unit and be detected. Peptide digestion and liquid chromatography further characterize solvent-accessible protein sites. When applied to Aβ structural dynamics, HDX-MS readily differentiated between monomers, protofibrils, and fibrils (Kheterpal et al., 2003). This study revealed that 40% of the backbone amide hydrogens were located within the core of protofibrils and that this increased to 60% in mature fibrils (Kheterpal et al., 2003), indicating dynamic changes of the positioning of residues throughout fibril formation. The same technique also demonstrated how the core of Aβ42 (residues 20-35) “seeded” aggregation, while the hydrophobic C-terminus played a minor but indispensable role (Zhang et al., 2013).
Single molecule force spectroscopy (SMSF) offers a profound resolution to study the mechanical stability of Aβ secondary structures. It consists of the molecule-of-interest attached to a surface with a probe on each end. The probe exerting large force is a sharp tip at the free end of a cantilever-beam for atomic force microscopy (AFM-SMSF; Eghiaian & Rico, 2014; Figure 3C). Apart from measuring forces applied to the molecule, changes in reflection angle of a laser beam off the surface of the cantilever can give information on topography (Eghiaian & Rico, 2014). Of note, AFM-SMSF, coupled with other methods, was instrumental in showing nucleation as a critical step in fibril formation (Harper, Lieber & Lansbury, 1997). This technique showed that Aβ1-40 β-sheets attached to one another in a reversible zipping motion (Kellermayer et al., 2005). Currently, high-speed AFM is emerging and has already contributed to elucidating the formation of false fibril branching points (Milhiet et al., 2010).
Researchers are constantly refining the above techniques and combining them for optimization. For instance, HDX was coupled with Raman spectroscopy for determining the Aβ peptide psi dihedral angles, the nearby environment of aromatic amino acids, and overall core structure of the fibril (Kurouski et al., 2015). Nevertheless, the sophisticated set-up, accessibility, and requirement of training make these experimental techniques better candidates for validation than discovery.
Simulations and Experiments go Hand in Hand in Understanding Aβ Aggregation
It is evident now that simulations and experimental approaches are complementary to each other. One could benefit from the atomistic resolution that MD simulations propose for Aβ fibrillation and aggregation. The experimental evidence was solid for the biological relevance of these models. The idea of validations across different platforms is typically employed in refining experimentally solved structures (Raval, Piana, Eastwood, Dror & Shaw, 2012; Schröder, 2015). It has been extended by, for example, coupling modelling with NMR or small-angle X-ray scattering (SAXS) to understanding fluctuations of protein structures (Bernadó et al., 2010; Yang et al., 2007). In the Aβ field, the integration of in vitro and in silico approaches has offered meaningful insights into Aβ aggregation.
One contribution of combining the two approaches concerns the role of water. Water has been considered critical since early on as some drew parallelism of fibril formation with crystallization: In the latter process, escape of water molecules stands as a necessity (Thirumalai, Reddy & Straub, 2012), which resembles fibril formation where the landscape of solvation must change to accommodate aggregation. In Aβ fibrils, this happens early on (Tarus, Straub & Thirumalai, 2006) and is probably related to the establishment of hydrophobic interactions that organise aggregation. Many experimental and simulation studies have captured extensive conformational plasticity mediated by different contexts of water. Simulations exhibited discrepancies with respect to the amount of water within the pre-fibril nuclei (Krone et al., 2008; Tarus, Straub, & Thirumalai, 2006). The simulations of water channels, e.g., around the Asp23-Lys28 salt bridge (Lemkul & Bevan, 2010; Tarus et al., 2006), were validated by 2D infrared spectroscopy as previously discussed (Kim et al., 2009). This provides an example of integrating the two approaches: simulations described interesting interactions between different parts of Aβ with water, while experimental techniques addressed the discrepancies among in silico models.
Integrating in silico and in vitro approaches also contributed to understanding the influence of membrane lipid composition in Aβ42 embedment at the membrane, which modulates Aβ aggregation. This has first been primed by “lipidomics” strategies, characterising the compositions and prevalence of membrane lipid species across the human brain (Svennerholm, Boström, Jungbjer & Olsson, 1994). Contemporary large-scale shotgun MS-based lipid measurements (Han, 2010) added to this, by demonstrating that cholesterol redistribution is dependent upon aging and AD. Computational approaches contributed to the mechanistic explanation of these phenomena, e.g. in a study by Liguori, Nerenberg, Paul, and Head-Gordon (2013). They showed, with free energy calculations, that the abundant extracellular cholesterol favours extrusion of N-terminals of Aβ fibrils, probing it to oligomerise and aggregate. On the other hand, the helicity at the C-terminal of Aβ peptides contributed to retain themselves at the plasma membrane. Amongst other intrinsic and delicate components of the local environment which “omics” experiments have primed, MD simulations give fine resolution into their role for mediating Aβ association, folding, and oligomerization.
It is also interesting to look at the interplay of computational and experimental approaches in a broader, time-lapse view of Aβ research. Long before simulations were available, conventional structural biology had established that cross-β structures were common in aggregating amyloid fibrils. ssNMR provided direct evidence that the same peptide sequence could form a vast variety of polymorphs (Figure 1A). Although oligomers existed before protofibrils and fibrils, the rearrangement of weaker oligomer intermediates might be directed to either fibril formation or growth into various stable forms of oligomers, depending on the physical and chemical environment (Figure 4i-iv) as concluded in a ssNMR study (Tay, Huang, Rosenberry & Paravastu, 2013).
Once the oligomers are committed to forming protofibrils and fibrils, they start to elongate extensively. In MD-simulated seeded growth (Kahler et al., 2013), single layers of protofibrils attempted to elongate until stability broke and they dissociated into short protofibrils. Later, pairing occured and resulted in protofibril pairs and possibly triplets with varying molecular structures caused by subtle differences in growth conditions, as shown by EM and ssNMR (Petkova et al., 2005). The increase in β-sheet structures gave the fibril greater stability as revealed by Fourier transform infrared spectroscopy (Kodali, Williams, Chemuru & Wetzel, 2010). Additionally, this allowed further elongation into large fibrils (Figure 4v-x).

Figure 4. Computational and experimental approaches propose an intergrative model of amyloid aggregation dynamics. The dynamic process of amyloid aggregation is summarized from research utilizing techniques of ssNMR, MD simulation, EM, Fourier transform infrared spectroscopy, kinetic assays, radio-labelling and cell viability experiments. (i-iii) Aβ monomers grow by opportunistic reversible adhesion to each other into different sizes of oligomers. (iv) Weak oligomers might undergo structural rearrangement into more stabilized oligomeric aggregates or (v) commit to extendible fibril nucleation. (vi) Short monolayer protofibrils elongate and (vii) fragment upon instability. (viii) Repeated growth and fragmentation generate short regular fragments which enable fibril thickening into varying layers and morphology, e.g., two-layer striated-ribbons or three-layer twisted morphology as illustrated, depending on physiological conditions. (ix) These structures continue to elongate with minor unit detachment, and branch out by secondary nucleation. (x) Sustained growth gives massive fibril meshes.
Subjected to the underlying molecular structures, these fibrils were polymorphic with unique physical properties. Nevertheless, all of these structures (including the oligomers) are in dynamic equilibrium with constant fragmentation and secondary nucleation which enable growing of new fibrils along the old ones, according to a series of kinetic, radio-labelling and cell viability experiments (Cohen et al., 2013). Altogether, this elaborate pathway of Aβ aggregation is the product of combining computational and experimental approaches in discovering biophysical basis at each step. Either one of these is indispensable in advancing mechanistic understanding of aggregation.
CONCLUSION AND FUTURE PERSPECTIVES
We presented an overview of the mechanism of Aβ aggregation and highlighted that computational and experimental approaches go hand in hand behind this. The synergy has contributed to identifying the distinct steps of aggregation as well as their biophysical underpinnings. This has allowed numerous attempts in identifying the so-called “wonder drugs”, which effectively antagonise aggregation and bring the least side effects (Karran et al., 2011). An understanding of the aggregative structural polymorphs can lead to possible strategies to target Aβ aggregation, e.g. by utilising the agents that disrupt salt bridges to destabilise the fibrils. For the theoretical biophysicists, the success of detailing the aggregative process also bears significant implications. It is this well-positioned biophysical problem that puts MD simulation to its best use, probing details that in vitro approaches often lack. The rise of simulation as a well-recognised approach to study biomolecular dynamics, as well as the excitement that the distributed computing project Folding@home (http://folding.stanford.edu/home) has brought to both the academic field and the media, bring protein folding and misfolding research to new heights. Further refinement, e.g., using patient-derived fibrils, would resolve ambiguities in this mechanism, considering the model in Figure 1B(iii) being the only, to our knowledge, published patient-derived Aβ fibril structure (Paravastu, Qahwash, Leapman, Meredith & Tycko, 2009). This would inspire further computational simulation and experimental validation to provide details on the basis of preferring one polymorph over another and their relevance to neuropathology. More broadly, combining different lines of investigations help greatly in understanding mechanistic aspects of many critical proteins in other diseases awaiting research efforts.
REFERENCES
Ahmed, M., Davis, J., Aucoin, D., Sato, T., Ahuja, S., Aimoto, S., Elliott, J. I., Van Nostrand, W. E., & Smith, S. O. (2010). Structural conversion of neurotoxic amyloid-β(1-42) oligomers to fibrils. Nature Structural & Molecular Biology, 17(5), 561-567.
Arnaud, C. H. (2009). Tools for Amyloids. Chemical and Engineering News, 87.
Bernadó, P., Modig, K., Grela, P., Svergun, D. I., Tchorzewski, M., Pons, M., & Akke, M. (2010). Structure and Dynamics of Ribosomal Protein L12: An Ensemble Model Based on SAXS and NMR Relaxation. Biophysical Journal, 98(10), 2374-2382.
Bruker Optics Inc. (2013). Study of protein conformation with FTIR. In Bruker Optics Inc (Ed.), (pp. 1-3). USA.
Cerasoli, E., Ryadnov, M. G., & Austen, B. M. (2015). The elusive nature and diagnostics of misfolded Aβ oligomers. Frontiers in Chemistry, 3, 17.
Cohen, S. I. A., Linse, S., Luheshi, L. M., Hellstrand, E., White, D. A., Rajah, L., Otzen, D. E., Vendruscolo, M., Dobson, C. M., & Knowles, T. P. J. (2013). Proliferation of amyloid-β42 aggregates occurs through a secondary nucleation mechanism. Proceedings of the National Academy of Sciences, 110(24), 9758-9763.
Eghiaian, F., & Rico, F. (2014). High-Speed Atomic Force Microscopy: Imaging and Force Spectroscopy. FEBS Letters, 588(19), 3631-3638.
Eyles, S. J., & Kaltashov, I. A. (2004). Methods to Study Protein Dynamics and Folding by Mass Spectrometry. Methods, 34, 88-99.
Han, X. (2010). Multi-dimensional mass spectrometry-based shotgun lipidomics and the altered lipids at the mild cognitive impairment stage of Alzheimer's disease. Biochimica et Biophysica Acta (BBA) - Molecular and Cell Biology of Lipids, 1801(8), 774-783.
Harper, J. D., Lieber, C. M., & Lansbury, P. T. J. (1997). Atomic Force Microscopic Imaging of Seeded Fibril Formation and Fibril Branching by the Alzheimer's Disease Amyloid-β Protein. Chemistry and Biology, 4(12), 951-959.
Jernigan, R. L., & Bahar, I. (1996). Structure-derived potentials and protein simulations. Current Opinion in Structural Biology, 6(2), 195-209.
Kahler, A., Sticht, H., & Horn, A. H. C. (2013). Conformational Stability of Fibrillar Amyloid-β Oligomers via Protofilament Pair Formation - A Systematic Computational Study. PloS One, 8(7), 1-12.
Karran, E., Mercken, M., & De Strooper, B. (2011). The amyloid cascade hypothesis for Alzheimer's disease: an appraisal for the development of therapeutics. Nature Reviews Drug Discovery, 10(9), 698-712.
Kellermayer, M. S. Z., Grama, L., Karsai, A., Nagy, A., Kahn, A., Datki, Z. L., & Penke, B. (2005). Reversible Mechanical Unzipping of Amyloid β-Fibrils. Journal of Biological Chemistry, 280(9), 8464-8470.
Kheterpal, I., Lashuel, H. A., Hartley, D. M., Walz, T., Lansbury, P. T. J., & Wetzel, R. (2003). Aβ Protofibrils Possess a Stable Core Structure Resistant to Hydrogen Exchange. Biochemistry, 42(48), 14092-14098.
Kim, Y. S., Liu, L., Axelsen, P. H., & Hochstrasser, R. M. (2009). 2D IR provides evidence for mobile water molecules in β-amyloid fibrils. Proceedings of the National Academy of Sciences, 106(42), 17751-17756.
Klimov, D. K., & Thirumalai, D. (2003). Dissecting the Assembly of Aβ16-22 Amyloid Peptides into Antiparallel β Sheets. Structure, 11(3), 295-307.
Kodali, R., Williams, A. D., Chemuru, S., & Wetzel, R. (2010). Aβ(1-40) forms five distinct amyloid structures whose beta-sheet contents and fibril stabilities are correlated. Journal of Molecular Biology, 401(3), 503-517.
Krone, M. G., Hua, L., Soto, P., Zhou, R., Berne, B. J., & Shea, J. E. (2008). Role of Water in Mediating the Assembly of Alzheimer Amyloid-β Aβ16−22 Protofilaments. Journal of the American Chemical Society, 130(33), 11066-11072.
Kurouski, D., Van Duyne, R. P., & Lednev, I. K. (2015). Exploring the Structure and Formation Mechanism of Amyloid Fibrils by Raman Spectroscopy: a Review. Analyst, 140(15), 4967-4980.
Lemkul, J. A., & Bevan, D. R. (2010). Assessing the Stability of Alzheimer’s Amyloid Protofibrils Using Molecular Dynamics. Journal of Physical Chemistry. B, 114(4), 1652-1660.
Liguori, N., Nerenberg, Paul S., & Head-Gordon, T. (2013). Embedding Aβ42 in Heterogeneous Membranes Depends on Cholesterol Asymmetries. Biophysical Journal, 105(4), 899-910.
Milhiet, P. E., Yamamoto, D., Berthoumieu, O., Dosset, P., Le Grimellec, C., Verdier, J. M., Marchal, S., & Ando, T. (2010). Deciphering the Structure, Growth and Assembly of Amyloid-like Fibrils Using High-speed Atomic Force Microscopy. PloS One, 5(10), 13240.
Näslund, J., Schierhorn, A., Hellman, U., Lannfelt, L., Roses, A. D., Tjernberg, L. O., Silberring, J., Gandy, S. E., Winblad, B., & Greengard, P. (1994). Relative abundance of Alzheimer Aβ amyloid peptide variants in Alzheimer disease and normal aging. Proceedings of the National Academy of Sciences, 91(18), 8378-8382.
Paravastu, A. K., Qahwash, I., Leapman, R. D., Meredith, S. C., & Tycko, R. (2009). Seeded growth of β-amyloid fibrils from Alzheimer's brain-derived fibrils produces a distinct fibril structure. Proceedings of the National Academy of Sciences, 106(18), 7443-7448.
Perez, A., Morrone, J. A., Simmerling, C., & Dill, K. A. (2016). Advances in free-energy-based simulations of protein folding and ligand binding. Current Opinion in Structural Biology, 36, 25-31.
Petkova, A. T., Leapman, R. D., Guo, Z., Yau, W. M., Mattson, M. P., & Tycko, R. (2005). Self-Propagating, Molecular-Level Polymorphism in Alzheimer's β-Amyloid Fibrils. Science, 307, 262-265.
Raval, A., Piana, S., Eastwood, M. P., Dror, R. O., & Shaw, D. E. (2012). Refinement of protein structure homology models via long, all-atom molecular dynamics simulations. Proteins: Structure, Function and Bioinformatics, 80(8), 2071-2079.
Robustelli, P., Stafford, K. A., & Palmer, A. G. (2012). Interpreting Protein Structural Dynamics from NMR Chemical Shifts. Journal of the American Chemical Society, 134(14), 6365-6374.
Schröder, G. F. (2015). Hybrid methods for macromolecular structure determination: experiment with expectations. Current Opinion in Structural Biology, 31, 20-27.
Schwierz, N., Frost, C. V., Geissler, P. L., & Zacharias, M. (2016). Dynamics of Seeded Aβ40-Fibril Growth from Atomistic Molecular Dynamics Simulations: Kinetic Trapping and Reduced Water Mobility in the Locking Step. Journal of the American Chemical Society, 138(2), 527-539.
Shirahama, T., & Cohen, A. S. (1967). High-resolution electron microscopic analysis of the amyloid fibril. Journal of Cell Biology, 33(3), 679-708.
Svennerholm, L., Boström, K., Jungbjer, B., & Olsson, L. (1994). Membrane Lipids of Adult Human Brain: Lipid Composition of Frontal and Temporal Lobe in Subjects of Age 20 to 100 Years. Journal of Neurochemistry, 63(5), 1802-1811.
Tarus, B., Straub, J. E., & Thirumalai, D. (2006). Dynamics of Asp23−Lys28 Salt-Bridge Formation in Aβ10-35 Monomers. Journal of the American Chemical Society, 128(50), 16159-16168.
Tay, W. M., Huang, D., Rosenberry, T. L., & Paravastu, A. K. (2013). The Alzheimer's amyloid-β(1-42) peptide forms off-pathway oligomers and fibrils that are distinguished structurally by intermolecular organization. Journal of Molecular Biology, 425(14), 2494-2508.
Thirumalai, D., Reddy, G., & Straub, J. E. (2012). Role of Water in Protein Aggregation and Amyloid Polymorphism. Accounts of Chemical Research, 45(1), 83-92.
Tomic, J. L., Pensalfini, A., Head, E., & Glabe, C. G. (2009). Soluble fibrillar oligomer levels are elevated in Alzheimer's disease brain and correlate with cognitive dysfunction. Neurobiology of Disease, 35(3), 352-358.
Yang, L. W., Eyal, E., Chennubhotla, C., Jee, J., Gronenborn, A. M., & Bahar, I. (2007). Insights into Equilibrium Dynamics of Proteins from Comparison of NMR and X-Ray Data with Computational Predictions. Structure, 15(6), 741-749.
Yu, L., Edalji, R., Harlan, J. E., Holzmann, T. F., Lopez, A. P., Labkovsky, B., Hillen, H., Barghorn, S., Ebert, U., Richardson, P. L., Miesbauer, L., Solomon, L., Bartley, D., Walter, K., Johnson, R. W., Hajduk, P. J., & Olejniczak, E. T. (2009). Structural Characterization of a Soluble Amyloid β-Peptide Oligomer. Biochemistry, 48, 1870-1877.
Zhang, Y., Rempel, D. L., Zhang, J., Sharma, A. K., Mirica, L. M., & Gross, M. L. (2013). Pulsed Hydrogen-Deuterium Exchange Mass Spectrometry Probes Conformational Changes in Aβ Peptide Aggregation. Proceedings of the National Academy of Sciences, 110(36), 14604-14609.