
Authors: Greg D. Owens, Patrice D. Smith, Nafisa M. Jadavji
Institution: Department of Neuroscience, Carleton University, 1125 Colonel By Dr., Ottawa, ON, K1S 5B6
Date: December 2016
doi: 10.22186/jyi.31.6.39-43
ABSTRACT
The primary pathway for removing homocysteine, a potentially neurotoxic molecule, from circulation is via 5-methyltetrahydrofolate. This molecule is converted from folate via an enzyme known as methylenetetrahydrofolate reductase (MTHFR). Polymorphisms in the MTHFR gene have been linked to various pathologies (e.g. neurological disease) and animal models have been developed to study the in vivo effects of the deficiency. These models have revealed increased levels of apoptosis in the cerebellum and hippocampus and potential modulation of neurogenesis, which may contribute to the pathologies viewed. The aim of this study was to evaluate neurogenesis during late development. Brain tissue from 3-week-old male mice with different MTHFR genotypic was collected and stained for phosphohistone H3 (PH3) and 4',6-diamidino-2-phenylindole (DAPI). Cell counts were performed in order to quantify PH3 positive cells in the hippocampus (dentate gyrus), cerebellum and cortex. There were no significant differences between genotype groups in all areas analysis. This result suggests that MTHFR status does not affect neurogenesis levels at this particular stage of development. Future research should consider the potential for confounding methylation pathways, such as the betaine-mediated mechanism, and investigating the effect on earlier developmental periods.
INTRODUCTION
Folate metabolism is a key mechanism in the brain that allows the downstream alteration of a variety of proteins and plays a role in the synthesis of nucleotides (Kamen, 1997). These folate-mediated effects are necessary for the production of new neural cells and thus are essential to the overall health of the brain during development, adulthood and aging (McGarel, Pentieva, Strain & McNulty, 2015). One mechanism through which folates affect protein function is through the initial methylation of homocysteine to methionine. Homocysteine is a cytotoxic molecule when in high levels that produces a variety of negative effects, including endoplasmic reticulum stress, excitatory amino acid receptor over-activation, kinase hyperactivity and DNA damage (Ho, Ortiz, Rogers & Shea, 2002). These effects have been associated with many clinical pathologies in humans, being indicated as a contributing factor to cognitive impairment (Almeida et al., 2005), neural tube defects (Felkner, Suarez, Canfield, Brender & Sun, 2009), brain atrophy (den Heijer et al., 2003), stroke (Hankey & Eikelboom, 2001) and cardiovascular disease (Frosst et al., 1995; Wierzbicki, 2007). Thus, the ability for folates to methylate homocysteine to its nontoxic derivative methionine plays a large role in protecting against neurotoxicity. However, the links between these illnesses and homocysteine are still not fully understood, with many downstream biochemical pathways still needing to be discovered. Research into increased homocysteine levels and altered folate metabolism confirms a variety of cytotoxic effects in Caenorhabditis elegans (Ortbauer et al., 2016), Drosophila melanogaster (Blatch, Stabler & Harrison, 2015) and Sacchaomyces cerevisiae (Kumar et al., 2011). Thus, the need to utilize mammal models of increased levels of homocysteine is necessary to produce potential theories of illness.
One such model looks at the knockout of a particular enzyme in the homocysteine cycle, known as methylenetetrahydrofolate reductase (MTHFR). Folate itself cannot directly methylate homocysteine, thus it must first be converted from the form it is ingested to its primary circulating form, 5-methyltetrahydrofolate (5-methyl-THF). The key enzyme to this process is the aforementioned MTHFR, which catalyzes the production of 5-methyl-THF from a less abundant form 5,10-methyl-THF, which then methylates homocysteine. Thus, this enzyme is essential to both the metabolism of folate and homocysteine. This cycle is highlighted in Figure 1.
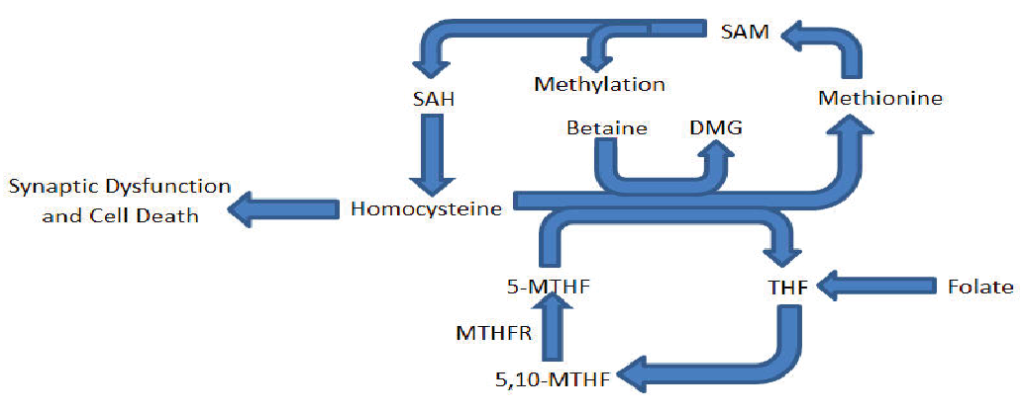
Figure 1. Partial diagram of the methionine/homocysteine and folate cycle outlining key contributing molecules to this research.
The occurrence of MTHFR deficiency is not uncommon in humans, with two common mutations producing reduced or lack of function. One of these deficiency-causing mutations is homozygous in approximately 18 percent of humans (Zittan et al., 2007). As many as 34 mutations in this gene, however, have been identified in individuals with homocystinuria, a genetic condition resulting in elevated levels of homocysteine that is associated with neurological and vascular problems (Leclerc, Sibani & Rozen, 2000). Studies into MTHFR in animal models have confirmed its role in preventing homocysteine-mediated neurotoxicity and associated pathologies (Chen et al., 2001; Chen, Schwahn, Wu, He & Rozen, 2005; Jadavji et al., 2012). Some of these pathologies include changes to motor control, mood and cognitive function, all of which have some connection to the altered morphology of the hippocampus and cerebellum specifically (Jadavji et al., 2012). The combination of the modelled results and the clinical implications of this gene thus offer significant intrigue into the role of folates in protecting against neurotoxicity.
Many of the associated pathologies, including cognitive impairment, brain atrophy and neural tube defects would suggest that the production of new and fully-functioning neurons is altered in response to high levels of homocysteine (Boot et al., 2003; Black, 2008). Neurogenesis, or the production of new neurons, occurs most prominently prenatally across species (Clancy, Darlington & Finlay, 2001, and is tightly linked to overall brain and cognitive function (Siwak-Tapp et al., 2007). Neurogenesis is also inherently linked with apoptosis, or cell death, as during development, neural pathways are trimmed via apoptosis. Additionally, adult neural death must be replaced by new cells in order to maintain the function of that brain region. Adult neurogenesis almost exclusively occurs in the dentate gyrus of the hippocampus and the olfactory bulb (Ming & Song, 2011), although there is research suggesting adult cell proliferation in the cerebellum (Ponti, Peretto & Bonfanti, 2008) and the cortex (Gould, Reeves, Graziano & Gross, 1999). As a result of this, it is of great interest to study the effect of increased homocysteine levels on the rates of neurogenesis in these regions to determine if blunted neurogenesis occurs as a result of increased homocysteine levels and if this mediates any of the observed pathologies.
Previous research has already begun to look in some of these regions with regards to neurogenesis and homocysteine levels. Research conducted by Jadavji et al. in 2012, used adult MTHFR knockout mouse models to test the association between neurogenesis and hyperhomocysteinemia-mediated pathologies. The results showed that homozygous knockouts for MTHFR had severely decreased cognitive functioning in object recognition tests, while apoptosis levels in the dentate gyrus were also elevated in the same genotype (Jadavji et al., 2012). This would suggest that without cell replacement in the dentate, cognitive function is greatly reduced, as a result of elevated homocysteine.
Similarly, research into other areas of the brain associated with neurogenesis appears to produce comparable results. In 2005, Chen, Schwahn, Wu, He & Rozen analyzed cell death levels in the cerebellum of MTHFR knockout mice, and found that homozygous knockouts also showed increased apoptosis of cerebellar neurons in both the intragranular and extragranular layers during the first two weeks of postnatal development (Chen, Schwahn, Wu, He & Rozen, 2005). As previously mentioned, pruning of neural pathways occurs during this time frame and alterations to cell death would result in changes to cerebellar patterning and could potentially contribute to neuropathology.
Thus, in order to determine if the pathologies viewed as a result of elevated homocysteine levels are partially mediated by alterations to neurogenesis levels, this study aimed to analyze the levels of neurogenesis in three of the regions associated with adult cell proliferation; the dentate gyrus, the cerebellum and the cortex. Analysis of neurogenesis in these regions in MTHFR knockout mice was used to determine if and propose how neurogenesis reductions may mediate homocysteine-related neuropathology.
MATERIALS AND METHODS
Animal Experimentation
All experiments were approved by Montreal Children’s Hospital Animal Care Committee in accordance to the Canadian Council on Animal Care guidelines. The mice used were from the C57B1/6 genetic background. The breeding of each status of MTHFR has been described previously (Jadavji et al., 2012). Mice were fed standard mouse chow (Envigo) and water ad libitum. They were sacrificed at 3 weeks from male mice.
Immunofluorescence
Slides of sagittal-oriented brain tissue from 3-week-old animals were deparaffinized in fresh xylene twice for 5 minutes each. They were then rehydrated in absolute alcohol for 5 minutes, followed by reduced concentration alcohol gradually at 95, 80 and 70% alcohol for 3 minutes each. The slides were then rinsed using PBS twice for 5 minutes each before Antigen Unmasking Solution was used for 20 minutes at 95°C to retrieve antigens on the slides. 10 μg/mL Proteinase K in Tris/EDTA solution was used to perform cell permeabilization for 10 minutes at room temperature before 5% goat serum in PBS blocked non-specific binding sites in the samples for 30 minutes. This allows for the targeted antibody-antigen interaction to occur. These slices were then stained using 1:100 concentration rabbit anti-phospho histone H3 mitosis marker (PH3; Cell Signalling) as the primary antibody to identify cells that were proliferating overnight at 4°C. Alexa Fluor (Cell Signalling) antibody at a dilution of 1:200 was then used for 40 min at room temperature. 4',6-diamidino-2-phenylindole (DAPI) was used as a general stain for the visualization of all cells and section were coverslipped with Vectashield Hardset Mounting Media in order to preserve the staining and were stored at 4°C.
Immunofluorescence Analysis
The stained slides were imaged using Infinity Analyze software, where an image under fluorescence for PH3 and DAPI were captured for each of the regions of interest; the dentate gyrus of the hippocampus, the cerebellum and the cortex. The images for each fluor respectively, were combined using Image J software (NIH) and the final combined images were manually quantified for co-localization of the two immunofluorescent tags. As the cerebellum and cortex were larger and unable to be imaged all in one photo, three separate lobes were imaged for each cerebellar sample, and three sections from each cortex (anterior, medial and posterior) and the counts were averaged from every sample for each specimen into one mean per mouse.
Statistical Analysis
Data was analyzed using SPSS and GraphPad software. One-way analysis of variance (ANOVA) was conducted on the three levels of MTHFR in each of the regions of interest, with p < .05 set as the significance threshold.
RESULTS
Previous research into MTHFR knockout mice would suggest that neurogenesis would likely be reduced in the mice with lost function of the enzyme. Previous research by Chen et al. (2001) confirmed the knockout status of MTHFR of this strain of mice, and can be referred to for Western blot results. As the goal of the research was to determine if MTHFR status had an impact on neurogenesis in the 3 week old mice, the co-localization of DAPI and PH3 staining was compared to determine if there was any difference between genotype groups.
Dentate Gyrus
First, in the dentate gyrus (Figure 2), the three statuses of MTHFR produced mean co-localized cell counts of 4.10 (n = 4, SD = 1.49) for the wild-type, 4.33 (n = 3, SD = 4.04) for the heterozygotes and 4.50 (n = 4, SD = 1.78) for the full knockouts. There was no difference between genotype groups (Figure 2(C), F(2,8) = 0.03, p = 0.974).

Figure 2. Combined images of the co-localization of PH3 and DAPI immunofluorescence staining in the dentate gyrus of a (A) wild-type MTHFR mouse (+/+) and (B) homozygous knockout MTHFR mouse (-/-). 200X magnification. Scale bar 50µm. Mean co-localization count in the dentate gyrus of MTHFR knockout mice and controls (C). No statistically significant differences were viewed (F(2,8) = 0.03, p = 0.974). Standard deviations are represented by the standard deviation bars attached to each column.
Cerebellum
Next, the same measures were taken for the cerebellum (Figure 3). This procedure produced a mean co-localized cell count for the wild-type mice of 9.30, (n = 5,SD = 4.51), 8.55 (n = 6, SD = 4.49) for the MTHFR heterozygotes and 5.64 (n = 5, SD= 1.52) for the complete knockouts. The analysis of variance showed no difference between groups (Figure 3(C), F(2,13) = 1.29, p = 0.309).

Figure 3. Combined images of the co-localization of PH3 and DAPI immunofluorescence staining in the cerebellum of (A) a wild-type MTHFR mouse (+/+) and (B) a homozygous knockout MTHFR mouse (-/-). 200X magnification. Scale bar 50µm. Mean co-localization count in the cerebellum of MTHFR knockout mice and controls (C). No statistically significant differences were viewed (F(2,13) = 1.29, p = 0.309). Standard deviations are represented in the figure by the standard deviation bars attached to each column.
Cortex
Finally, the levels of co-localized cells were recorded for the cortex of the mice, as imaged in Figure 4. The mean co-localized cell count for the wild-type mice was 8.06 (n = 4, SD = 1.93), 6.19 (n = 4, SD= 3.71) for the heterozygous knockouts and 7.42 (n = 5, SD= 4.62) for the complete knockouts. There was no difference between groups (Figure 4(C), F(2,10) = 0.26, p = .773).

Figure 4. Combined images of the co-localization of PH3 and DAPI immunofluorescence staining in the cortex of (A) a wild-type MTHFR mouse (+/+) and (B) a homozygous knockout MTHFR mouse (-/-). 200X magnification. Scale bar 50µm. Mean co-localization count in the cortex of MTHFR knockout mice and controls (C). No statistically significant differences were viewed (F(2,10) = 0.26, p = .773). Standard deviations are represented in the figure by the standard deviation bars attached to each column.
DISCUSSION
This research produced no statistically significant changes in the co-localization counts of PH3 and DAPI in any of the three main regions of interest, the dentate gyrus, cerebellum and cortex in response to MTHFR status. This would suggest that the relative level of MTHFR does not have a significant impact on neurogenesis at this point of development in male mice, and is not an ideal candidate for explaining altered neurogenesis and neuropathology seen in response to increased homocysteine. Despite this unexpected result, there are numerous reasons why this study may have failed to reject the null hypotheses.
The first reason may simply have to do with a small sample size. The levels of MTHFR had a range of n-values of 3 to 6, thus even after procedures to reduce variance by calculating a mean for each mouse before the genotype mean, there was still a high level or variance for most statuses. This high variance greatly contributes to the failure to reject the null hypothesis of the one-way ANOVAs, especially for the cerebellum which had the highest F-ratio. If the sample size was increased for each MTHFR level, it is possible that the resulting reduced variance could lead to a statistically significant effect. Procedural issues were not the only potential source of confound producing this result. As mentioned in the introduction, mice are generally into their “adolescent” period by three weeks, and thus have completed the majority of their developmental neurogenesis (Clancy, Darlington & Finlay, 2001). As no significant change in neurogenesis was viewed at this stage of development, it could perhaps be beneficial to look at younger mice that are still going through developmental neurogenesis. It is possible that earlier neurogenesis may be more sensitive to MTHFR status, suggested by the many studies showing developmental neurological defects, especially in the neural tube (Felkner, Suarez, Canfield, Brender & Sun, 2009). Future research should consider looking at mice earlier in neurological development for alterations to neurogenesis as a result of MTHFR status.
In addition to changing the age of the mice used, another possible reason for viewing this result could be an alternative method of methylating homocysteine. Betaine is well established as a supplementary methyl donor for the homocysteine to methionine conversion known to reduce homocysteine concentrations (Olthof & Verhoef, 2005). Derived from choline, betaine is regularly produced in the body through the catalytic activity of two enzymes, choline dehydrogenase (CHDH) and betaine aldehyde dehydrogenase (BADH). Betaine is then converted into its methyl-donating form via betaine homocysteine methyltransferase (BHMT). Thus, these results viewed may show that at this key stage of development there is an alternative method of reducing homocysteine and neuroprotection in response to MTHFR deletion. This method could involve the upregulation of any of the CHDH, BADH or BHMT enzymes, resulting in increased betaine-mediated methylation of homocysteine, preventing the neurotoxic effects. This theory is not completely hypothetical, as previous research mentioned by Chen, Schwahn, Wu, He & Rozen, in 2005, showed that betaine augmentation reduced the effect of MTHFR deletion in cerebellar development, the region closest in this study to having statistically significant effects (Chen, Schwahn, Wu, He & Rozen, 2005). Although the supplementation of that study came from an external source, it could be valuable to assess the relative transcript levels of betaine-related enzymes in future research of folate metabolism and the homocysteine cycle.
It may also be of value in future research to sample levels of methyl marks on methylated acceptors in these regions of interest. As methionine can be metabolized to S-adenosylmethionine, protein methylation would be expected to be viewed in mice with effective folate metabolism. This could potentially provide a valuable measure of whether homocysteine was being converted to methionine at regular levels or not, perhaps confirming confounding effects from betaine.
MTHFR has continuously been indicated in numerous pathologies and has been modelled effectively as a knockout paradigm. Age of the mice and confounding enzymatic pathways should both be considered in future research into the models. Despite no statistically significant results here, this study still contributes essential information into the mechanisms of neurogenesis and folate metabolism.
REFERENCES
Almeida, O. P., Flicker, L., Lautenschlager, N. T., Leedman, P., Vasikaran, S. & van Bockxmeer, F. M. (2005). Contribution of the MTHFR gene to the causal pathway for depression, anxiety and cognitive impairment later in life. Neurobiology of Aging, 26(2), 251-257. doi:10.1016/j.neurobiolaging.2004.03.007.
Black, M. M. (2008). Effects of vitamin B12 and folate deficiency on brain development in children. Food and Nutrition Bulletin, 29(2): S126-S131.
Blatch, S. A., Stabler, S. P. & Harrison, J. F. (2015). The effects of folate intake on DNA and single-carbon pathway metabolism in the fruit fly Drosophila melanogaster compared to mammals. Comparitive Biochemistry and Physiology: Part B Biochemistry and Molecular Biology, 189, 34-39. doi: 10.1016/j.cbpb.2015.07.007.
Boot, M. J., Steegers-Theunissen, R. P., Poelmann, R. E., Van Iperen, L., Lindemans, J. & Gittenberger-de Groot, A. C. (2003). Folic acid and homocysteine affect neural crest and neuroepithelial cell outgrowth and differentiation in vitro. Developental Dynamics, 227(2): 301-308. doi: 10.1002/dvdy.10303
Chen, Z., Karaplis, A. C., Ackerman, S. L., Pogribny, I. P., Melnyk, S., Lussier-Cacan, S., Chen, M. F., Pai, A., John, S. W., Smith, R. S., Bottiglieri, T., Bagley, P., Selhub, J., Rudnicki, M. A., James, S. J. & Rozen, R. (2001). Mice deficient in methylenetetrahydrofolate reductase exhibit hyperhomocysteinemia and decreasedmethylation capacity, with neuropathology and aortic lipid deposition. Human Molecular Genetics, 10(5), 433-443.
Chen, Z., Schwahn, B. C., Wu, Q., He, X. & Rozen, R. (2005). Postnatal cerebellar defects in mice deficient in methylenetetrahydrofolate reductase. International Journal of Developmental Neuroscience, 23, 465-474. doi:10.1016/j.ijdevneu.2005.05.007.
Clancy, B., Darlington, B. & Finlay, B. L. (2001). Translating developmental time across mammalian species. Neuroscience, 105, 7-17.
den Heijer, T., Vermeer, S. E., Clarke, R., Oudkerk, M., Koudstaal, P. J., Hofman, A. & Breteler, M. M. (2003). Homocysteine and brain atrophy on MRI of non-demented elderly. Brain, 126, 170-175.
Felkner, M., Suarez, L., Canfield, M. A., Brender, J. D. & Sun, Q. (2009). Maternal serum homocysteine and risk for neural tube defects in a Texas-Mexico border population. Birth Defects Research, 85(6), 574-581. doi: 10.1002/bdra.20545.
Frosst, P., Blom, H. J., Milos, R., Goyette, P., Sheppard, C. A., Matthews, R. G., Boers, G. J., den Heijer, M., Kluijtmans, L. A., van den Heuvel, L. P. & Rozen, R. (1995). A candidate genetic risk factor for vascular disease: A common mutation in methylenetetrahydrofolate reductase, Nature Genetics, 10, 111-113.
Gould, E., Reeves, A. J., Graziano, M., S. & Gross, C. G. (1999). Neurogenesis in the neocortex of adult primates. Science, 15(286), 548-552. doi: 10.1126/science.286.5439.548.
Hankey, G. J. & Eikelboom, J. W. (2001). Homocysteine and stroke. Current Opinion in Neurology, 14, 95-102.
Ho, P. I., Ortiz, D., Rogers, E. & Shea, T. B. (2002). Multiple aspects of homocysteine neurotoxicity: Glutamate excitotoxicity, kinase hyperactivation and DNA damage. Journal of Neuroscience Research, 70(5), 694-702. doi: 10.1002/jnr.10416.
Jadavji, N. M., Deng, L., Leclerc, D., Malysheva, O., Bedell, B. J., Caudill, M. A. & Rozen, R. (2012). Severe methylenetetrahydrofolate reductase deficiency in mice results in behavioral anomalies with morphological and biochemical changes in hippocampus. Molecular Genetics and Metabolism, 106, 149-159. doi:10.1016/j.ymgme.2012.03.020.
Kamen, B. (1997). Folate and antifolate pharmacology. Seminars in Oncology, 24(5 Suppl 18), 30-39. PMID: 9420019.
Kumar, A., John, L., Maity, S., Manchanda, M., Sharma, A., Saini, N., Chakraborty, K. & Sengupta, S. (2011). Converging evidence of mitochondrial dysfunction in a yeast model of homocysteine metabolism imbalance. The Jounral of Biological Chemistry, 286(24), 21779-21795. doi: 10.1074/jbc.M111.228072.
Leclerc D, Sibani S, Rozen R. Molecular Biology of Methylenetetrahydrofolate Reductase (MTHFR) and Overview of Mutations/Polymorphisms. Austin (TX): Landes Bioscience; 2000.
McGarel, C., Pentieva, K., Strain, J. J. & McNulty, H. (2015). Emerging roles for folate and related B-vitamins in brain health across the lifecycle. The Proceedings of the Nutrition Society, 74(1), 46-55.
Olthof, M. R. & Verhoef, P. (2005). Effects of betaine intake on plasma homocysteine concentrations and consequences for health. Current Drug Metabolism, 6, 15-22.
Ortbauer, M., Ripper, D., Fuhrmann, T., Lassi, M., Auernigg-Haselmaier, S., Sitegler, C. & Konig, J. (2016). Folate deficiency and voer-supplemntation causes impaired folate metabolism: Regulation and adaption mechanisms in Caenorhabditis elegans. Molecular Nutrition & Food Research, 60(4), 949-956. doi: 10.1002/mnfr.201500819.
Ponti, G., Peretto, P. & Bonfanti, L. (2008). Genesis of neuronal and glial progenitors in the cerebellar cortex of peripuberal and adult rabbits. PLoS One, 3(6), e2366. doi:10.1371/journal.pone.0002366.
Siwak-Tapp, C. T., Head, E., Muggenburg, B. A., Milgram, N. W. & Cotman, C. W. (2007). Neurogenesis decreases with age in the canine hippocampus and correlates with cognitive function. Neurobiology of Learning and Memory, 88(2), 249-259. doi:10.1016/j.nlm.2007.05.001.
Wierzbicki, A. S. (2007). Homocysteine and cardiovascular disease: A review of the evidence. Diabetes and Vascular Disease Research, 4, 143-150.
Zittan, E., Preis, M., Asmir, I., Cassel, A., Lindenfeld, N., Alroy, S., Halon, D. A., Lewis, B. S., Shiran, A., Schliamser, J. E. & Flugelman, M. Y. (2007). High frequency of vitamin B12 deficiency in asymptomatic individuals homozygous to MTHFR C677T mutation associated with endothelial dysfunction and homocysteinemia. American Journal of Physiology: Heart and Circulatory Physiology, 293(1), 860-865. doi: 10.1152/ajpheart.01189.2006.