

Author: Christina Sweeney
Institution: Louisiana State University
Date: July 2007
Abstract:
The research that will be presented in this paper concerns the synthesis and testing of a bimetallic homogenous catalytic system based on a tetraphosphine ligand system. The [Rh2H2(µ -CO)2(rac-et,ph-P4)]2+ homogeneous catalyst system discovered by G. G. Stanley's laboratory can be resolved into pure enantiomers and has been shown to be one of the best asymmetric hydroformylation catalysts for vinyl esters with 85% enantiomeric excess (ee) and 4:1 branched to linear regioselectivity for vinyl acetate (Stanley 2005).
The procedure involves four different aspects: ligand synthesis, separation of racemic- and meso-et, ph-P4 ligand, preparation of the dirhodium catalyst, and hydroformylation catalysis. I successfully synthesized the binucleating tetraphosphine ligand and dirhodium catalyst precursor and studied bimetallic cooperativity in hydroformylation catalysis. For future research, we hope to perform additional catalytic runs, compare the catalyst to other monometallic rhodium catalysts, and to determine better reaction conditions for the hydrogen producing aldehyde-water shift catalysis.
Introduction:
The research that will be presented in this paper is concerning the synthesis and testing of the dirhodium catalytic system. The research involves four different aspects: ligand synthesis, separation of racemic- and meso-et, ph-P4 ligand, preparation of the dirhodium catalyst precursor, and hydroformylation catalysis.
The synthesis of the tetraphosphine ligand that is the foundation for the dirhodium catalyst system is a fairly long and complicated process. My goal was to become proficient with it and to be able to complete it without any assistance. The future resolution of racemic-et, ph-P4 ligand into pure R,R- and S,S- enantiomers will use a chiral preparatory HPLC column and an automatic collection HPLC system to isolate the two enantiomers of the tetraphosphine ligand.
The dirhodium catalyst preparation step is very simple as it only involves reacting the racemic-et,ph-P4 ligand with two equivalents of [Rh(nbd)2](BF4) (nbd = norbornadiene) to make the catalyst precursor, [Rh2(nbd)2(et,ph-P4)](BF4)2. This reaction is usually close to quantitative in yield. The last step is the catalytic testing. GC (and sometimes NMR) is used to determine the product distributions. Previous runs were done using pure acetone solvent, but Professor Stanley's group has discovered that an acetone/water solvent mixture (30% water by volume) is far more effective at dramatically reducing catalyst degradation reactions and, increasing both the rate and overall selectivity of the hydroformylation catalysis.
Homogenous and Heterogeneous Catalysis:
Heterogeneous catalysis is where the catalyst is in a different phase (solid, liquid, and gas) relative to the reactants and products, and also provides a surface for the chemical reaction to take place on. Homogeneous catalysis is where the catalyst is a discrete molecular species in the same phase as the reactants, typically in solution. Homogeneous catalytic systems have several advantages relative to heterogeneous catalytic systems. These advantages include: far more selective for a single product, far more active, more easily studied from chemical and mechanical aspects due to the solution state, and more easily modified for optimizing selectivity and activity. The main disadvantages of homogeneous systems are that they are far more sensitive to permanent deactivation and more difficult in achieving product/catalyst separation. Despite the advantages of homogeneous systems in relation to heterogeneous systems, approximately 95% of chemical and petrochemical industries use heterogeneous catalytic systems (Stanley 2005).
In determining the efficiency of a homogeneous or heterogeneous catalyst, there are a few factors that chemists look at: the turnover frequency (TOF) or, the total number of turnovers performed before catalyst deactivation, selectivity for products, and the reaction conditions. The number of turnovers is very effective in rating catalysts the higher the number the better the catalyst. The turnover frequency (TOF) states the number of passes through the catalytic cycle per unit of time. This number is usually determined by taking the number of moles of product produced, dividing that by the number of moles of catalyst used in the reaction, and then dividing that by the time to produce the given amount of product (Stanley 2005). The units, therefore, are usually just time-1.
Hydroformylation (Oxo) Catalysis:
Hydroformylation, or oxo catalysis, is one of the most important homogeneous industrial processes in use today. The reaction involves the reaction of alkenes, carbon
monoxide, and hydrogen to produce aldehyde products (Stanley 2005). As seen above in Figure 1, hydroformylation can give two aldehyde products. The products will be either a linear or branched (iso) aldehyde; however the linear is generally preferred by industry for bulk commodity chemicals (Stanley 2005).

Figure 1: Standard Example of a Hydroformylation Reaction
Asymmetric Bimetallic Hydroformylation:
Hydroformylation produces two isomeric products: the linear aldehyde and the branched aldehyde. In the case of asymmetric hydroformylation the branched products have a new chiral center alpha to the carbonyl of the aldehyde for most alkenes. For most industrial applications, the linear aldehyde product is the desirable form of the aldehyde. However, branched aldehydes and related compounds are desirable in pharmaceutical and fine chemical markets, assuming that one can control the chirality (Barker 2005). An example is the pain reliever naproxen (also known as Aleve). (R)-naproxen is a deadly liver toxin, while (S)-naproxen has the desired anti-inflammatory characteristics. Asymmetric hydroformylation (Figure 2) is one way in which (S)-naproxen could be selectively synthesized.

Figure 2: Steps of the Hydroformylation Reaction for (S)-naproxen
A desirable asymmetric hydroformylation catalyst would produce highly-branched regioselectivity along with enantioselectivity (ee). The ee is defined as the absolute value of the difference of the S enantiomer from the R enantiomer, divided by the sum of the two enantiomers, which is then multiplied by 100. A catalyst that makes an equal amount of R and S enantiomers has 0% ee. 85% or higher is generally considered a good ee, depending on what the best known catalyst can do relative to that being reported.
Rhodium Catalysis:
The [Rh2H2(-CO)2(rac-et,ph-P4)]2+ homogeneous catalyst system discovered by G. G. Stanley's laboratory can be resolved into pure enantiomers and has been shown to be one of the best asymmetric hydroformylation catalysts for vinyl esters with 85% enantiomeric excess (ee) and 4:1 branched to linear regioselectivity for vinyl acetate (Stanley 2005). The homogeneous catalyst system is based on a binucleating tetraphosphine ligand (racemic- and meso-et,ph-P4) that can both bridge and chelate two transition metal centers, producing bimetallic complexes that only have a single, conformationally flexible bridging group.
Materials and Methods:
The entire process of making the catalyst is actually a four-step process. It involves the synthesis of the ligand (shown in figure 3 below), separation of racemic and meso-et, ph-P4 ligand, dirhodium catalyst precursor preparation, and hydroformylation catalysis. Each step will be discussed in detail concerning the actual reactions and techniques used.

Figure 3: The overall reaction for the synthesis of the racemic and meso-et, ph-P4 ligand
Ligand Synthesis:
The ligand is made from "bridge", Ph(H)PCH2P(H)Ph, and vinyldiethylphosphine, which both have to be made in separate steps. The bridge is made in one step, while the vinyldiethylphosphine requires a two-step synthesis.
Ph(H)PCH2P(H)Ph, "Bridge" synthesis (Courtney 2004):
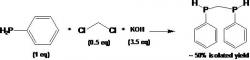
article_1115_order_9
The first step is to weigh a 500 mL Schlenk flask for the reaction and product collection and a 250 mL Schlenk flask for the KOH solution. Using a drybox, weigh out CH2Cl2 (0.5 eq) and PhPH2 (1 eq), then add both to a Schlenk flask with a stirbar, close with septum, and remove from box. Degas DMF via N2 sparging (100 mL for 20g PhPH2) and transfer to prepared Schlenk flask via canula. Cool flask in ice bath.
Next, prepare the saturated KOH (aq) solution in degassed deionized H2O (3.5 eq KOH). Add the solution drop wise via canula after cooling solution in ice bath for 30 minutes. While flasks are in ice bath, add KOH to the stirred DMF solution. Remove the ice baths and allow reaction to warm to reaction temperature with stirring. Should stir at least two hours under N2, however can stir overnight.
After solution turns completely colorless with white precipitate, add approximately 100 mL degassed deionized H2O to react with any remaining PhPH2 and dissolve solid KCl. Remove the bridge via degassed pentane extraction (canula transfers) into the preweighed Schlenk flask. Remove the solvent via vacuum and warm bridge to 80o C for approximately one hour in a hot water bath with vacuum to remove any impurities. Do no heat above 85o C. Add hexane to bridge and filter through a Schlenk frit to remove oxidized bridge, and then remove hexane via vacuum.
Diethylchlorophosphine (Courtney 2004):

article_1115_order_10
In glove box, charge a Schlenk flask with PCl3, a stir bar, and an equal volume of t-glyme. Use a large graduated cylinder to make pouring and measuring easier and use the same graduated cylinder for t-glyme as for PCl3 to rinse out remaining PCl3. Close flask with septum. In glove box, charge a second Schlenk flask with t-glyme from a different graduated cylinder (equal volume as intended for ZnEt2). Then quickly and carefully measure and add ZnEt2 using same graduated cylinder. Close flask with septum. (ZnEt2 is highly air sensitive and may smoke slightly). Remove the flasks from glove box and cool in ice bath for at least 30 minutes. With vigorous stirring in PCl3 flask, add ZnEt2 drop-wise via canula at the rate of approximately 2 drops per second. Keep ice baths cold. After addition is complete, use trap-to-trap distillation to collect product (shown in Figure 4 below).
Flush N2 through reaction flask while quickly adding the trap-to-trap apparatus with the Teflon valve open (to allow N2 flow) and then quickly close the Teflon valve. Attach a clean preweighed Schlenk flask to receiving end. Keeping the Teflon valve closed, attach trap-to-trap stopcock to vacuum and evacuate receiving flask. Cool receiving flask in liquid N2 bath. With both N2 stopcocks closed, open the Teflon valve. Slowly and carefully open vacuum stopcock to bring reaction flask to boiling. Do not allow reaction to bump" into the trap-to-trap apparatus. Heat reaction flack to approximately 80o C in a warm water bath to get the entire product to distill over. Continue heating until solution stops boiling. After distillation is complete, fill system with N2 and allow receiving flask to thaw. After thawed, flush N2 through receiving flask while you disconnect it and put a septum in place. Store in freezer.
Vinyldiethylphosphine (Courtney 2004):
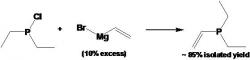
article_1115_order_11
In the glove box put 100 mL 1M VinylMgBr in THF (clear brown liquid) into a 500 mL two-necked Schlenk flask. Add 100 mL t-glyme and a stir bar into the 500 mL schlenk flask. It is very important that the vinylMgBr solution (in THF) is clear of any precipitate. Add a trap-to-trap to the middle neck flask and a septum to the side neck. Be sure the Teflon valve is closed. In the glove box charge a 250 mL Schlenk flask with ClPEt2 (12 mL 0.09 mol) and then put 100 mL of t-glyme. Close the flask with a septum. Remove both flasks from the glove box. To the receiving end of the trap-to-trap apparatus, attach a tiny flask simply to close off the system.
Apply vacuum to the vinylMgBr solution via the vacuum stopcock and remove the THF and collect it in the line trap. Weigh the THF to determine the volume of THF removed. Must remove at least 95 mL. Heat the vinylMgBr solution to approximately 80oC with a warm water bath and open the Teflon valve. After solvent exchange is complete, fill the vinylMgBr flask with N2 and close Teflon valve. Cool both the vinylMgBr and the ClPEt2 solutions in ice baths for approximately 30 minutes. Then, add the ClPEt2 via canula drop-wise in the stirring reaction flask. Collect the vinyldiethylphosphine via a trap-to-trap distillation into a clean preweighed Schlenk flask. Keep the reaction below 85oC, because t-glyme may distill over at 90oC.
et,ph-p4 Ligand (Courtney 2004):

article_1115_order_12
Under nitrogen atmosphere, combine one equivalent of bridge to 2.2 equivalents of vinyldiethylphosphine in a small (less than 50 mL) Schlenk flask with a stir bar. Expose to the Xenon arc lamp while stirring for at least 8 hours, but longer exposure times may ensure complete reaction. Test for complete reaction via 31PNMR (solution in hexanes). All bridge must have reacted! If unreacted bridge remains, continue UV exposure until all bridge has reacted. The addition of more vinyldiethylphosphine is okay, but should not be necessary if excess was originally used. After all of bridge has reacted, remove excess vinyldiethylphosphine by applying vacuum to the reaction flasks for approximately one hour, while heating the flask to about 75oC by a warm water bath. Save the collected vinyldiethylphosphine for future reactions (it should be pure).
Partial Separation of racemic and meso et,ph-P4:
Separate the racemic and meso mixture by adding hexane to the solution and placing it in the freezer overnight. Once the meso ligand forms white precipitate, remove the racemic ligand from the meso ligand via canula and place in another flask. Remove excess hexane under reduced pressure; add more hexane to the solution and place in freezer. The process is continued until mixture is > 80% racemic.
Catalyst Precursor:
The ligand is part of the catalyst precursor, which will be combined with other compounds to make what we believe is the active catalyst system [Rh2H2(µ -CO)2(rac-et,ph-P4)]2+. The catalyst precursor is shown in Figure 5 below.
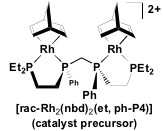
Figure 5: The molecular structure of the catalyst precursor, et, ph-P4
Synthesis of Rh(nbd)(acac) (Barker 2005):
Add Rh(CO)2(acac) and norbornadiene to a 250 mL schlenk flask. Attach a reflux condenser to the flask and heat while stirring in an oil bath at 90oC for three hours. After reaction is complete, cool, filter, and remove the unreacted norbornadiene by vacuum. Use 10 mL of THF and the heat gun to dissolve the powder and then add 3 or 4 seconds of hexane. Filter using a fritted funnel, cap funnel, and place crystals in the glove bow freezer overnight.
Synthesis of [Rh(nbd)2](BF4) (Barker 2005):
Dissolve Rh(nbd)(acac) in 30 mL of THF in the glove box. Cool solution to -20oC by placing in freezer. After solution is cooled, add HBF4 OEt2 drop wise. Add norbornadiene via a canula into the reaction flask. Place flask in freezer to cool to 20oC for two hours. Collect the precipitate by filtration.
Synthesis of [rac-Rh2(nbd)2(et,ph-P4)](BF4)2 (Barker 2005):
Dissolve the [Rh(nbd)2](BF4) in 10 mL of CH2Cl2 in a 250 mL flask. Dissolve the rac-et,ph-P4 in 5 mL of CH2Cl2. Add the rac-et,ph-P4 drop wise to the [Rh(nbd)2](BF4) with the solution stirring for 30 minutes. Remove the CH2Cl2 under vacuum. Heat the red-orange solid slightly and add acetone drop wise (be careful the pressure might pop off the septum). Put in freezer overnight and allow the solid to crystallize.
hydroformylation Runs:
In figure 6 below, it shows two diastereotopic forms, racemic and meso, which are yielded from the synthesis of the ligand.
Place 1mM of [rac-Rh2(nbd)2(et,ph-P4)] in 80 mL 30% water/acetone in an autoclave. The autoclave is heated to 90°C and the catalyst solution soaks under H2/CO gas (90 psig) for 20 min with stirring (1000 rpm). 1 M 1-hexene is injected after soaking, and data is collected until olefin is consumed. Samples are collected during and at the end of runs for GC analysis.

Figure 6: The diasteromeric, racemic and meso, et,ph-P4 ligands
Results:
In the synthesis of the bridge, we used 15.093 grams PhPH2, 5.832 grams CH2Cl2 (DCM), and 26.27 grams KOH. When making the bridge there is a color and state change during the addition of the KOH to the DMF solution, the reaction will turn bright yellow and from a solid precipitate. The oxidized bridge will be present as a solid, but the desired Ph(H)PCH2P(H)Ph bridge will be a clear colorless liquid after the hexane is removed. The molecular weights for the compounds in the reaction are as follows: CH2Cl2 is 89.83 g/mol, KOH is 56.11 g/mol, and the Ph(H)PCH2P(H)Ph bridge is 232.20 g/mol. The expected chemical shifts for the 31P NMR of the rac- and meso- Ph(H)PCH2P(H)Ph bridge are -53 ppm and -54 ppm and Figure 7 shows that there are indeed chemical shifts at -53.1 ppm and -54.1 ppm. The expected percent yield is approximately 40-50%, however we got a slightly better than usual percent yield of 52%.

Figure 7: The 31P NMR of the Ph(H)PCH2P(H)Ph bridge shows chemical shifts (peaks) at -53.1 ppm and -54.1 ppm for the racemic and meso diastereomers, which assists in identifying the compounds in the bridge.
In the synthesis of the diethylchlorophosphine, we used 21.0 grams PCl3 in 30mL of t-glyme and 20.071 grams ZnEt2 in 35mL of t-glyme. The ClPEt2 is a clear colorless liquid, which should be stored in the freezer. The molecular weights for the compounds in the reaction are as follows: PCl3 is 137.5 g/mol, ZnEt2 is 123.5 g/mol, and ClPEt2 is 124.55 g/mol. The expected chemical shift for the 31P NMR is 112 ppm, however small amounts of the unknown impurity with a 66 ppm chemical shift is considered okay. The expected percent yield for this reaction is approximately 70% and we got an isolated yield of 83%.
In the synthesis of the vinyldiethylphosphine, we used 15.758 grams ClPEt2 and 140.0 mL of vinylmagnesium bromide, H2C=CHMgBr. When the t-glyme is added to the vinylMgBr, a white precipitate forms in the flask. When the t-glyme is added to the 250 mL ClPEt2 charged Schlenk flask the solution becomes clear and colorless. When the ClPEt2 is added to the reaction flask there is a color change to light yellow and a white precipitate forms. The vinyldiethylphosphine is a clear colorless liquid with a molecular weight of 116.14 g/mol. The expected chemical shift for the 31P NMR is 18 ppm and Figure 8 shows a chemical shift at -17.8 ppm. The expected percent yield for the reaction is 85+%, however it will contain some THF that cannot be easily separated. We got an isolated yield of 75%.

Figure 8: The 31P NMR of the vinyldiethylphosphine sample shows a chemical shift (peak) at -17.804 ppm, which assists in identifying the compounds in the vinyldiethylphosphine.
When photolyzing the vinyldiethylphosphine and the bridge to prepare the final et,ph-p4 ligand, the reaction mixture becomes more viscous as the reaction proceeds. When testing to determine if all of the bridge has reacted, the expected chemical shift for the bridge in the 31P NMR is 53 ppm and 54 ppm and vinyldiethylphosphine at 18 ppm. The mixed meso and racemic et,ph-p4 ligand is a colorless oil with a molecular weight of 464.49 g/mol. The expected chemical shifts for the 31P NMR are as follows: the external phosphine arms are around 17 ppm, the internal methylene-bridged racemic phosphine resonances are at 25 ppm, and the meso bridge phosphines are at 26 ppm. Figure 9 below shows that the external phosphines are at -15.8 ppm and -16.7, the racemic bridge is at -24.2 ppm, -24.5 ppm, and -24.9 ppm, and the meso bridge is at
-25.1. These chemical shifts are very close to the expected results.

Figure 9: The 31P NMR of the racemic- and meso-et,ph-p4 ligand sample shows chemical shifts (peaks) at -25.1 ppm, -24.9 ppm, -24.5 ppm, -24.2 ppm, -16.7 ppm and -15.8 ppm, which assists in identifying the compounds in the racemic and meso ligand.
When making Rh(nbd)(acac), the solution will turn from dark green to bright yellow. After removing any unreacted norbornadiene, the solution will solidify into a yellow powder, which crystallized to form yellow crystals. The expected yield for the reaction is 90-95%. The expected chemical shifts for the 1H NMR (CDCl3) are as follows: 1.22.0 (m, CH2 of nbd and CH3 of acac), 3.8-4.0 (m, CH of nbd), 5.3 (s, CO-CH-CO of acac), 6.2 and 6.7 (br s, and br m, olefinic CH of nbd).
In the making of [Rh(nbd)2](BF4), HBF4 OEt2 is added drop-wise which causes a color change from yellow to dark red. The addition of norbornadiene caused an orange-red precipitate to form. The expected percent yield for the reaction is 90-95%. The expected chemical shifts for the 1H NMR (CD2Cl2) are as follows: 1.7 (br s, CH2 of nbd), 4.3 (br s, CH of nbd), 5.3 and 5.6 (br m, and br s, olefinic CH of nbd).
In the final step in the dirhodium catalyst precursor synthesis, the red-orange solid will crystallize to yield orange crystals. In the synthesis of the catalyst, we used 2.075 grams [Rh(nbd)2](BF4) and 1.287 grams of the racemic-et,ph-p4 ligand. The expected percent yield for the reaction is 88-95%. The expected chemical shifts for the 31P NMR (CD2Cl2) are as follows: 47.5 (dm, JP-Rh = 156 Hz, internal phosphorus atoms) and 58.0 (dd, JP-P = 23 Hz and JP-Rh = 150 Hz, external phosphorus atoms). The expected chemical shifts for the 1H NMR (CD2Cl2) are as follows: 0.8-1.4 (m, PCH2CH3), 1.5-2.1 (m, PCH2CH3 and m, PCH2CH2P and s, CH2 of nbd), 2.9 (t, PCH2P), 3.6-4.2 (br d, CH of nbd), and 4.8 and 5.3 (br s, olefinic CH of nbd).
Discussion and Conclusion:
The purpose of the research was to introduce me to air-sensitive organometallic synthetic procedures, organophosphine and transition metal chemistry, and catalysis concepts that I had no prior knowledge. Due to the research, I became proficient in the inert atmosphere synthetic techniques, and a variety of laboratory equipment and was able to complete most of the project with minimal assistance. I successfully synthesized the binucleating tetraphosphine ligand and dirhodium catalyst precursor and studied bimetallic cooperativity in hydroformylation catalysis. This is supported by the data I collected and presented in this paper. Also, the purity of the ligand and catalyst precursor sample and the high percent yields support the successful synthesis.
The last thing I was supposed to do this summer was a hydroformylation run; however I was unable to complete this due to equipment malfunctions and time constraints. I plan on completing the runs when the school semester starts and the new machinery arrives. Theoretically the catalyst should run very well, since the NMRs of all the compounds used in the catalyst were at the expected chemical shifts and percent yields.
For future work, I plan on studying the bimetallic cooperativity in hydroformylation catalysis and to tackle the asymmetric project. I also want to compare the catalyst to other monometallic rhodium catalysts. Lastly, I want to do in situ FT-IR and NMR catalytic studies where I attempt to isolate more species in catalytic cycle and optimize the reaction conditions for the aldehyde-water shift catalysis.
References:
"Barker, BL (2005) Synthesis of Rh(nbd)(acac). Separation and in Situ Catalytic Testing of a Dirhodium Tetraphosphine Catalyst: 80."
"Barker, BL (2005) Synthesis of [Rh(nbd)2](BF4). Separation and in Situ Catalytic Testing of a Dirhodium Tetraphosphine Catalyst: 80-81."
"Barker, BL (2005) Synthesis of [rac-Rh2(nbd)2(et,ph-P4)](BF4)2. Separation and in Situ Catalytic Testing of a Dirhodium Tetraphosphine Catalyst: 81."
"Barker, BL (2005) Asymmetric Hydroformylation and Tetraphosphine Ligand Separations: Introduction. Separation and in Situ Catalytic Testing of a Dirhodium Tetraphosphine Catalyst: 16."
"Courtney, B (2004) Preparation of Bridge. Research Notes: 11."
"Courtney, B (2004) Preparation of Diethylchlorophosphine. Research Notes: 12-13."
"Courtney, B (2004) Preparation of Vinyldiethylphosphine. Research Notes: 14."
"Courtney, B (2004) Preparation of et,ph-P4 ligand. Research Notes: 15."
"Stanley, GG (2005) Catalysis Intro. Organometallic Chemistry: Lecture Notes: 4-8."
"Stanley, GG (2005) Hydroformylation. Organometallic Chemistry: Lecture Notes: 1 and 21."