

Author: Julie Hatfield
Institution: Juniata College
Date: October 2005
Abstract
The role of advanced glycation end-products (AGEs) in the development of complications in individuals with insulin-dependent diabetes mellitus (IDDM) has been explored by previous studies. However, the relationship between these reactive AGEs and diabetic complications are still somewhat unknown. Glycation (nonenzymatic glycosylation) processes, also known as the Maillard reactions, are a series of reactions between carbohydrates and free amino groups of proteins. The preliminary intermediates, (Amadori products; 1-amino, 1-deoxy, 2-ketoses), ultimately result in the formation of AGEs. AGEs in humans have been predominantly chemically characterized by the detection of pentosidine and N-carboxy-methyl lysine (CML). Both pentosidine and CML have been found to accumulate in skin and lens collagen matrix at accelerated rates in diabetic patients. Indications are that collagen in IDDM patients undergoes widespread chemical alterations that result in decreased solubility, alter binding affinities to enzymes, increased stability, accelerated cross-linking and increased browning. Accumulation of AGEs with structural alterations result in altered tissue properties that contribute to the reduced susceptibility to catabolism and to the aging of tissues. Also, when accelerated by hyperglycemia, AGE accumulation is believed to contribute to the gradual development of diabetic complications. Pentosidine concentrations in the skin of IDDM patients are often elevated and correlate to the severity of complications. It has also been suggested that pentosidine is not just a subset of diabetic complications but rather a general diagnostic feature of the disease process.
Introduction
The pathogenesis of diabetic complications continues to be a central issue in current diabetes research (15). One of the most prevalent metabolic syndromes world-wide, diabetes mellitus (DM), is characterized by hyperglycemia resulting in short-term metabolic changes in lipid and protein metabolism and long-term irreversible vascular and connective-tissue changes. These changes include diabetes-specific complications such as retinopathy, nephropathy and neuropathy and complications of the macro-vasculature such as atherosclerosis; potentially resulting in heart disease, stroke and peripheral vascular disease (11). Links between chronic hyperglycemia and the development of long-term diabetic-specific complications have been discovered and are yet not completely understood (11, 23).
Glycation, a chemical modification of proteins with reducing sugars, indicates a possible explanation for the association (7, 15) between hyperglycemia and the wide variety of tissue pathologies. Research suggests that reducing sugars can react with the amino groups of long-lived proteins to produce non-enzymatic cross-links (19, 23). Formations of these cross-links occur as end-stage products of the Maillard reaction; they are known as advanced glycation end-products (AGEs) (7, 20).
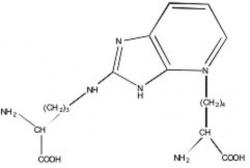
Figure 1. Structure of fluorophore P (Pentosidine).
AGEs are a class of complex, often unstable, reactive compounds formed in excess during aging and diabetes mellitus (23). According to the "glycation hypothesis," accumulation of AGEs alters the structural properties of tissue proteins and reduces their susceptibility to catabolism (7, 23). It has been shown that the process of AGE formation is accelerated by hyperglycemia (4, 7, 16). Some of the protein alterations observed in diabetic patients resemble those in much older, non-diabetic patients, suggesting diabetes induced early aging' (20).

Figure 2. Structure of carboxyl methyl lysine (CML).
The chemical nature of AGEs in vivo is largely unknown, but there is a growing population of structurally-defined AGE adducts such as pyrraline, pentosidine, N-carboxy-methyl lysine (CML) and crossline that are found to be elevated in diabetic tissues (7, 15, 21). The best found chemically characterized AGEs in humans are pentosidine and CML (see Figures 1, 2) (16, 23). Some of the highest levels of pentosidine have been detected in individuals afflicted with DM (19). Evidence has shown that elevated skin pentosidine levels in individuals with DM correlate with the severity of the complications (19). Initial investigations have shown that pentosidine can be detected in smaller levels in various tissues of noncollagenous origin, including the blood and the human lens (19).
Pentosidine is a fluorescent crosslink with visible wave length fluorescence, making it easy to detect (15). Methods for synthesizing and detecting AGEs such as pentosidine have been proposed in various studies (7, 14, 19). Prevention of AGE-mediated cell toxicity has been proposed as a key strategy in preventing the onset of diabetic complications and some age-related pathology (21). This review will continue to analyze evidence that AGEs play a significant role in diabetic complications considering various anti-AGE therapeutic strategies that appear to reduce the severity and onset of complications (4, 10, 12, 13, 15, 23).
Long-Term Complications Due to AGEs
Protein glycation and AGE formation are accompanied by increased free radical activity that contributes to the bimolecular damage in diabetes (1, 13, 23). AGEs act as mediators and can initiate a wide range of abnormal responses in cells and tissues such as the inappropriate expression of growth factors, alterations in growth dynamics, accumulation of extra-cellular matrix and initiation of cell death (21, 23), through decreased solubility, elasticity and enzymatic affinities in long-living proteins such as collagen (8, 15).
A number of these chemical and physical skin changes occur in human skin collagen with age and appear to be accelerated in diabetes (12, 13). AGE cross-linking reactions in collagen contributes to diabetic circulatory complications such as vascular stiffening and myocardial dysfunction (22, 23). Although the mechanisms underlying the development of the complications of diabetes are not fully understood, there is now a consensus that hyperglycemia does play an important role in the development of retinopathy, nephropathy, neuropathy and joint stiffness (10, 13). For example, increased serum and tissue levels of AGEs due to a reduced removal by the kidneys have been evident in end-stage renal failure (23). In vitro and in vivo studies have shown that AGEs result in irreversible cross-links in long living matrix structural proteins such as type IV collagen, laminin and fibronectin (23).
Biochemistry of AGEs
The formation of AGEs is implicated by the pathogenesis of long-term complications of diabetes (3, 6). There appears to be two general pathophysiologic mechanisms by which hyperglycemia leads to irreversible tissue damage (4). Intracellular hyperglycemia can result by increased flux through different metabolic pathways, changing glomerular basement membrane. Increases in the polyol pathway activity results in metabolic changes too, consequence of decreasing levels of NADPH, gluthathione and myoinositol (4).
A major consequence of hyperglycemia is excessive nonenzymatic glycosylation of proteins (3, 4, 10), primarily due to long-term exposure to elevated glucose concentrations (12). Nonenzymatic glycation may be occurring, although at a much slower rate than that seen most in IDDM patients (3). Nonenzymatic protein glycation (Mallard Reaction) by glucose is a complex cascade of condensations, rearrangements, fragmentations and oxidative modifications (3). Glucose chemically attaches to proteins and nucleic acids without the aid of enzymes, increasing the formation of AGEs (4). These AGEs form on intra- and extracellular proteins, lipids, and nucleic acids, leading to the generation of protein fluorescene and the irreversible cross-links (1, 11, 15). The formation of AGEs requires the reaction of reducing sugars like glucose, fructose, galactose, mannose and ribose (22). Interestingly, glucose is among the least reactive of the common sugars, perhaps leading to its evolutionary selection as the principle free sugar in vivo (1, 22).
For a given protein, the extent of nonenzymatic glycosylation is determined by the sum of effects of a number of independently acting variables such as pH, temperature, protein concentration, etc. (4, 9). Glucose concentration and incubation time are the most clinically relevant variables affecting the extent of nonenzymatic glycosylation (4). Characteristic to diabetics, increased levels of glucose concentrations cause the level of accumulated Amadori products on proteins to rise (4).
Non-enzymatic glycosylaton is a common posttranslational modification of proteins in vivo, resulting from reactions between glucose and amino groups on proteins, this process is coined the "Maillard reaction" and results in the formation of AGEs (1, 8, 11, 15, 23).
The Maillard Reaction (Non-enzymatic glycosylation)
AGEs form via a non-enzymatic condensation reaction between reducing sugars and -amino group or N-terminal groups (7, 10, 15, 16, 21-23) via a neucleophilic addition with formation of a Schiff base (1, 4). The Schiff base rapidly reaches an equilibrium level in vivo, reflecting the surrounding glucose concentration.
The chemically unstable Schiff bases form relatively fast and are highly reversible (4, 21, 22). Over a period of weeks, a slow chemical rearrangement of the Schiff base occurs, resulting in the formation of stable yet highly reversible ketoamine (Amadori product), an initial reaction product and intermediate in the formation of AGEs (1-5, 21, 22). Amadori adduct formation is slower but much faster than the reverse reaction, leading to accumulation of Amadori glycation products on various proteins (4, 21, 22). Reactive AGE-forming intermediates can arise from oxidative reactions ("glycoxidation") of free sugar or from initial Schiff base condensation products with protein amino groups, rather than just from the "classical" Amadori rearrangement (3). The presence of AGE cross-links in collagen is suggested to contribute to the severity of diabetic complications (1, 6, 12) although the degree to how much these relate is unknown.
Adducts of Glycation

Figure 3. Structure formed by glycation of lysine residues in protein, fructoselysine (FL).
The chemical nature of important AGEs as they occur in vivo is largely unknown due to their heterogeneous and unstable nature; however, there is a growing population of structurally defined AGE adducts such as pyrraline, pentosidine and CML, all of which have been found at elevated levels in diabetics (1, 4, 21). The two most commonly measured AGEs are CML and pentosidine, which are glycoxidation products, formed by sequential glycation and oxidation reactions (3). The adduct formed by glycation of lysine residues in protein is termed fructoselysine (FL) (see Figure 3), and levels of FL in hemoglobin, plasma proteins, collagen, hair, lens and numerous other proteins in the body are also known to increase in proportion to the degree of hyperglycemia in diabetes (8).
Early studies of nonenzymatic glycosylation showed that this process ultimately gives rise to pigmented, fluorescent and glucose-derived protein crosslinks (4). These pigmented, fluorescent compounds have been used to study the relationship between AGE formation and various tissue pathologies (2, 17, 19). Along with the brown color, fluorescence is one of the qualitative properties of AGEs (22). The fluorescent AGE crosslink pentosidine was first isolated and identified from dura mater collagen and has been identified in vivo in skin collagen and plasma proteins of diabetic patients (3, 7, 22).
Pentosidine Formation and Significance
Pentosidine has been detected and measured in tissue proteins by chemical and chromatographic methods (2, 17, 19). Pentosidine is unique in that it can be formed by the reaction of lysine and arginine, forming a fluorescent crosslink with any of several carbohydrate precursors including glucose, ribose, ascorbic acid, and 3-deoxyglucosone (see Figure 2) (7, 22). The development of increased fluorescence of proteins in diabetes and aging is enhanced by oxidation reactions and carbohydrate or lipid-dependent processes (7). It has been proposed that AGEs such as pentosidine are in fact active intermediates in the cross-linking of proteins and formation of reactive oxygen species (2).
Pentosdine has been found in a variety of tissues of human origin including skin, tracheal cartilage, cortical bone, aorta, cardiac muscle, lung, liver, kidney, lens, red blood cells, and blood proteins (19). Pentosidine has been found to accumulate in the skin and lens at accelerated rates in diabetics (11, 15). Overall, correlations between skin pentosidine levels and the severity of long-term complications indicate that pentosidine parallels severity (19). Formation of elevated skin pentosidine levels in IDDM patients with severe complications, although unclear, has been associated to poorer metabolic control compared to those with less severe complications (19). Therefore, skin pentosidine would be formed primarily from glycated skin collagen and should reflect cumulative glycohemoglobin AIC values which are now a chemically accepted indicator of glycemic control (4, 7).
Preventative Measures

Figure 4. Aminoguanadine (AG), a structurally identified AGE inhibitor.
Poor metabolic control and other characteristics of IDDM result in diabetic nephropathy, neuropathy, retinopathy, atherosclerosis and difficulty in healing wounds (1). Preventative measures for clinical problems that may be the result of accelerated AGE production with IDDM include improvement of glycemic control (4, 10). Recent studies have shown that when the quality of patient control is good (for example, patients who maintain a normal blood-glucose level) the concentrations of the AGE-products, CML and pentosidine, are typically lower (18). However, recent clinical trials suggest that when complications are already present, improvement of glycemic control alone may not be sufficient to prevent the continued progression of these pathologic processes, potentially due to the irreversibility of AGE formation as well as poor clearance mechanisms.
AGE inhibitors
Due to detrimental effects of AGEs, researchers attempt to find inhibitors of the advanced glycation process (6).Brownlee et al. suggest that optimal future therapies to minimize tissue damage may require pharmacologic agents that directly interfere with the self-perpetuating component of hyperglycemia-initiated tissue damage (4). Aminoguanadine (AG) (see Figure 4), an inhibitor of advanced glycation reactions in vitro, has been found to inhibit the development of diabetic complications in animal models of diabetes. (4, 12). Booth et al. suggest that these inhibitors can potentially react as a hydrazine with carbonyls of Amadori intermediates or can hunt for reactive dicarbonyls through its guanidinium moiety. However, the mechanism of AGE formation is only partially understood, making it difficult to identify the precise chemical products responsible for in vivo damage and thus impede the development of specific inhibitors.
Summary
A major consequence of hyperglycemia is excessive non-enzymatic glycosylation of proteins resulting in various protein-protein cross-links and non-crosslinked structures (4, 10, 22). AGE products contribute to long term complications of IDDM patients (2, 4, 17, 19). With the increasing rate of occurrence of IDDM, it is important to increase knowledge about AGEs and AGE-inhibitors. Through research it may be possible and beneficial to find substances that can be used to decrease or predict the occurrence of long term complications of AGE formation to improve the quality and length of life for IDDM patients.
References
1. Ahmed, N. (2005) Advanced glycation end products,role in pathology of diabetic complications. Diabetes Research and Clinical Practice 67, 3-21.
2. Baynes J. et al. (1999) Role of oxidative stress in diabetic complications. Diabetes 48, 1-9.
3. Booth A. et al. (1997) In vitro kenetic studies of formation of antigenic advanced glycation end products (AGEs). Journal of Biological Chemistry 272, 5430-5437.
4. Brownlee, M. et al. (1984). Nonenzymatic glycosylation and the pathogenesis of diabetic complications. Annals of Internal Medicine 101, 527-537.
5. Chellan P, et al. (2001) Early glycation products produce pentosidine cross-links on native proteins. Journal of Biological Chemistry 276, 3895-3903.
6. Degenhardt, T. et al. (1999) Aminoguanidine inhibits albuminuria, but not the formation of advanced glycation end-prodcuts in skin collagen of diabetic rats. Diabetes research and clinical practice 43, 81-89.
7. Dyer, D.G. (1993) Accumulation of maillard reaction products in skin collagen in diabetes and aging. Journal of Clinical Investigation 91, 2463-2469.
8. Dunn, J. et al. (1989). Oxidation of glycation proteins: age-dependent accumulation of N-(carboxylmethyl)lysine in lens proteins. Biochemistry 28, 9464-9468.
9. Eble, A.S. et al. (1983) Nonenzymatic glucose and glucose-dependent crosslinking or protein. Journal of Biological Chemistry 10, 9406-9412.
10. Forbes J. M. et al. (2003). Role of advanced glycation end products in diabetic nephropathy. Journal of American Society of Nephrology 14, S254-S258.
11. Hudson, J. et al. (2002) Glycation and diabetes:The RAGE connection. Current Science 83, 1515-1521.
12. Kochakian, M. (1996) Chronic dosing with aminoguanidine and novel advanced glycosylation end product-formation inhibitors ameliorates cross-linking of tail tendon collagen in STZ-induced diabetic rats. Diabetes 45, 1694-1700.
13. Lyons, T. et al. (1991). Decrease in skin collagen glycation with improved glycemic control in patients with insulin-dependent diabetes mellitus. Journal of Clinical Investigation 87, 1910-1915.
14. Maurizio, S. et al. (1995). Role of advanced glycation end-products (AGE) in late diabetic complications. Diabetes Research and Clinical Practice 28, 9-17.
15. McCance, D. et al. (1993) Maillard reaction products and their relation to complications in insulin-dependent diabetes mellitus. Journal of Clinical Investigators 91, 2470-2478.
16. Price, D. et al. (2001) Chelating activity of adcanced glycation end-product inhibitors. Journal of Biological Chemistry 276, 48967-48972.
17. Sajithlal, G. et al. (1998) Advanced glycation end products induce crosslinking of collagen in vitro. Biochimica et biophyisica Acta 1407, 215-224.
18. Schiel, R. et al. (2003) Improvement of the quality of diabetes control and decrease in the concentration of AGE-products in patients with type 1 and insulin-treated type 2 diabetes mellitus studied over a period of 10 years (JEVIN). Journal of Diabetes and Its Complications 17, 90-97.
19. Sell, D. et al. (1991) Pentosidine: a molecular marker for the cumulative damage to proteins in diabetes, aging, and uremia. Diabetes/Metabolisim Reviews 7, 239-251.
20. Sensi, M. et al. (1995) Role of advanced glycation end-products (AGE) in late diabetic complications. Diabetes Research and Clinical Practice 28, 9-17.
21. Stitt, A.W. (2001) Advanced glycation: an important pathological even in diabetic and age related ocular disease. British Journal of Ophthalomol 85, 746-753.
22. Ulrich, P and Cerami A. (2001) Protein glycation, diabetes and aging. Recent Progress in Hormone Research 56, 1-22.
23. Wautier, J.L. and Guillausseau, P.J (2001) Advanced glycation end products, their receptors and diabetic angiopathy. Diabetes Metabolism 27, 535-542.