

JACOB OYLER, EMILY NIEC, JESICA PHELAN, ALI TAUCHEN, RAECHEL TITTOR, TODD T. ECKDAHL
Biology Department, Missouri Western State University, 4525 Downs Drive, Saint Joseph, MO 64507, United States
ABSTRACT
Metabolic engineers use technology and ways of thinking from traditional science and engineering disciplines as well as the emergent field of synthetic biology to genetically manipulate cellular metabolic pathways for applications in medicine, energy, the environment, and more. Two important challenges to successful metabolic engineering are how to optimize the metabolic output and how to make products that are toxic to cells. Our research addressed these challenges with the development of a new strategy for the selection of optimal genotypes for enzyme production and the use of microfluidic droplets for cell-free protein synthesis. We focused on a prototype metabolic reaction by which caffeine demethylase converts caffeine into theophylline, and used a ribozyme-riboswitch that cleaves itself upon binding theophylline as the basis for selection of genetic regulatory elements that most efficiently produce caffeine demethylase. We designed and constructed a ribozyme chain molecule that couples caffeine demethylase gene constructs to magnetic nanoparticles in microfluidic droplets so that constructs with optimal constructs can be separated from suboptimal ones with a magnet. Our results include demonstration of the theophylline-dependent self-cleavage of a ribozyme, successful construction of the ribozyme chain molecule, and improvement of an isothermal amplification method called recombinase polymerase amplification for use in microfluidic droplets. Although technical limitations prevented us from demonstrating the full functionality of our selection strategy, our proposal and results have important implications for metabolic engineering. Our observation of a reversed theophylline dependence of the ribozyme-riboswitch when it was coupled to MNPs informs the use of ribozymes as tools for synthetic biology.
INTRODUCTION
Introduced in the 1970s, genetic engineering changed the world with important applications such as the biosynthesis of human insulin by bacteria (Quianzon and Cheikh 2012), the production of genetically modified foods (Bawa and Anilakumar 2013), and the development of microbes that can perform bioremediation (Ojuederie and Babalola 2017). In the early 2000s, synthetic biology was developed as a new approach to genetic engineering that integrates advances in molecular biology methods with engineering concepts and practices (Hughes and Ellington 2017; Meng and Ellis 2020), with wide-ranging applications in areas such as medicine (Flores Bueso et al. 2018; Choe et al. 2020), biofuels (Kumar 2020), and environmental bioremediation (Jaiswal and Shukla 2020). Many of these applications require metabolic engineering, which is the process by which cells can be genetically reprogrammed to produce one or more enzymes that control desired metabolic pathways. The design of genetic circuits for use in metabolic engineering can be based on a rational approach that relies on a quantitative understanding of naturally occurring or synthetic gene expression control elements and of the impact of orthogonal metabolism on cell physiology. An alternative approach is to exploit evolution by producing a population of genetic circuits and using artificial selection to optimize the desired metabolic output (Bali 2019, Pontrelli et al. 2018, Eckdahl et al. 2015). Metabolic engineers sometimes seek to produce metabolites that are toxic to cells (Lu 2017) by using in vitro gene expression, in which a cellular extract is prepared that can support transcription and translation directed by synthetic genetic circuits.
In this paper, we describe our efforts to respond to the need for new in vitro selection methods for metabolic engineering. The challenge we chose to pursue is optimization of the conversion by caffeine demethylase of caffeine to an asthma medication called theophylline. Although theophylline is well tolerated by cells, we used microfluidic droplets in our plan so that it could be adapted for the production of metabolites that are toxic to cells. Our plan is to produce caffeine demethylase in gene expression units that vary with regard to the strengths of the transcriptional promoter and the ribosome binding site (RBS). The goal is to introduce a library of gene expression units into microfluidic droplets at a concentration that ensures that there is no more than a single genotype variant in each droplet. Isothermal DNA amplification will be used to amplify each individual genotype variant within its droplet. Gene expression will then occur in the droplets using an E. coli cellular extract capable of cell-free protein synthesis that is included in the droplets. Droplets are expected to convert caffeine into theophylline at different rates depending on their genotypes. The key to selection of droplets that have efficiently produced theophylline is a Ribozyme Chain Molecule (RCM) that we invented. As illustrated in Figure 1, Panel A, the RCM includes a ribozyme-riboswitch that self-cleaves upon binding theophylline. The 5’ end of the ribozyme-riboswitch is ligated to the 3’ end of a DNA linker with a 5’ thiol modification that allows RCM molecules to be coupled to magnetic nanoparticles (MNPs) derivatized with amino groups. The 3’ end of the ribozyme-riboswitch is ligated to a DNA spacer containing an internal glycol modification. Inside droplets, isothermal amplification will be used to increase the number of copies of the gene expression units, and the glycol medication will prevent DNA synthesis from producing an RNA-DNA hybrid using the ribozyme-riboswitch as a template. The isothermal amplification method we chose is recombinase polymerase amplification (RPA; Lobato and O’Sullivan 2018). Instead of relying on temperature-dependent denaturation and annealing, RPA achieves primer binding with a recombinase that facilitates the invasion of a double-stranded target DNA sequence by a single-stranded primer, aided by single-stranded DNA binding protein (SSB). Figure 1, Panel B shows the process by which microfluidic droplets are formed by using micrometer-scale channels to control the interaction of a stream of aqueous solution with streams of oil. The image on the right is of droplets with MNPs inside them. Figure 1, Panel C illustrates how a magnet can be used to separate “winner” genotypes that have been separated from MNPs after theophylline-dependent ribozyme-riboswitch cleavage from “loser” genotypes that have not. The image shows how a magnet can be used to draw the MNPs to the side of a test tube within seconds.
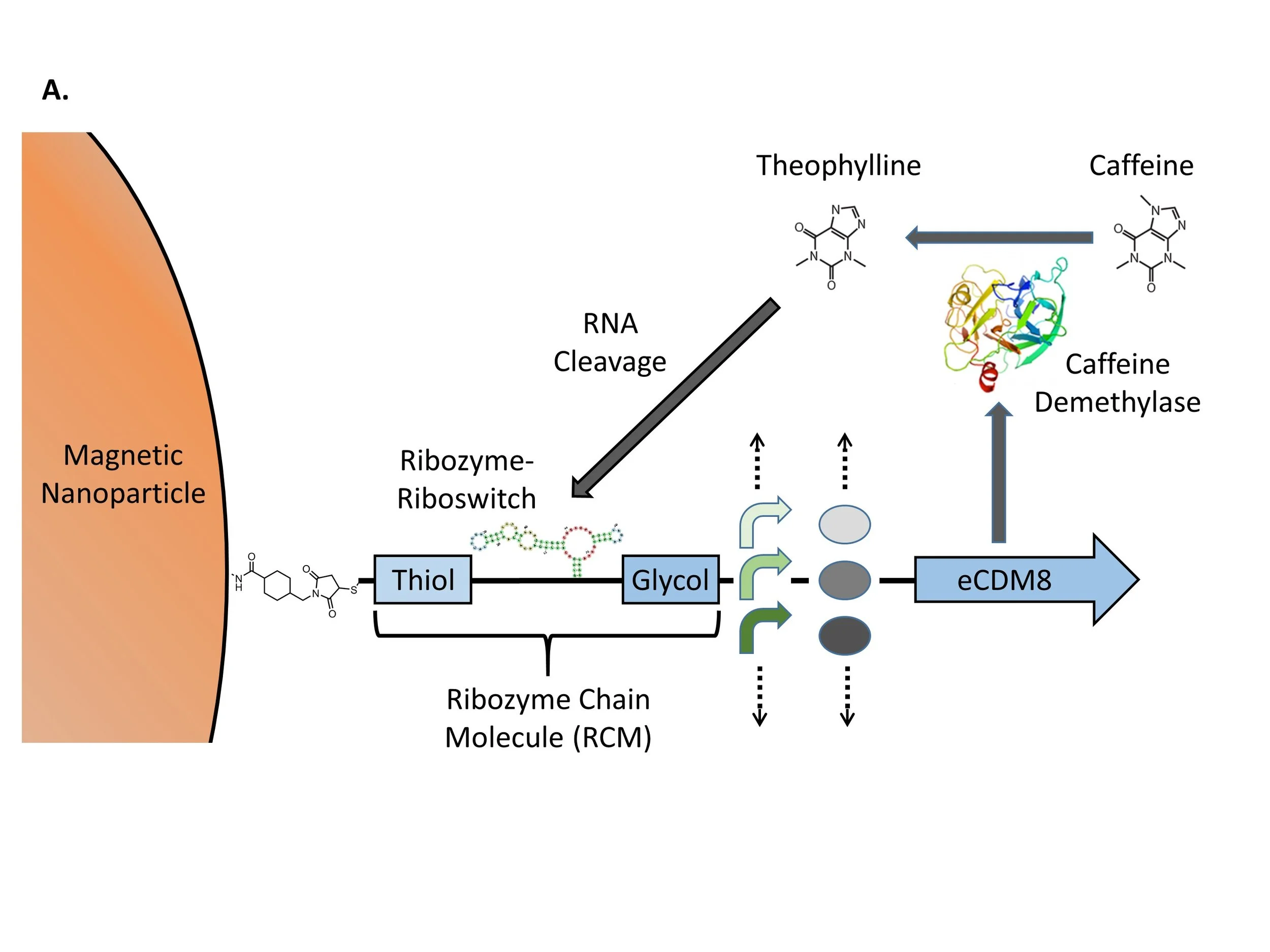

Figure 1. RCM Selection Strategy. A. Magnetic nanoparticles with amino derivatives are coupled to Ribozyme Chain Molecules (RCMs) that are composed of a thiol DNA linker (Thiol), the mammal ribozyme-riboswitch, and a glycol DNA spacer (Glycol). RCMs are used as primers to amplify a eCDM8 gene expression units that include a variable promoter and a variable ribosome binding site (RBS). B. MNP-RCM complexes are introduced into microfluidic droplets. The images show the PDMS droplet-generation microchip with aqueous and oil phases flowing and droplets produced with MNPs inside them. C. Selection is achieved by using a magnet to separate RCM genotypes that have been released from MNPs by theophylline-dependent ribozyme cleavage.
Our study has important implications for metabolic engineering. Our proposal to use a ribozyme-riboswitch as a basis for selection could be adapted for the optimization of other metabolic pathways by switching to other aptamer specificities. Our plans to use microfluidic droplets for the synthesis of products toxic to cells and to use RPA for isothermal amplification in droplets are also generalizable. We hypothesize that the theophylline dependence of the ribozyme-riboswitch will be retained in the context of an RCM coupled to MNPs. Evaluation of this hypothesis in the context of our results contributes to the use of ribozyme-riboswitches as tools for synthetic biology.
MATERIALS AND METHODS
Ribozyme-Riboswitch Synthesis and Testing
We gained access to the theophylline-dependent ribozyme-riboswitch sequence studied by Xiang et al. (Xiang et al. 2019) by ordering the following sequence from Integrated DNA Technologies (IDT) as a double-stranded DNA: 5’ GCTGTCACCGGAATACCAGCATCGTCTTGATGCCCTTGGAAGTCCGGTCTGATGAGTCCCATAAGGACGAAACAGC 3’. The sequence includes a promoter that enabled us to perform in vitro transcription using T7 RNAP Polymerase from New England BioLabs, Inc. (NEB). We purified the ribozyme-riboswitch RNA products of the reaction with the Monarch RNA Purification Kit from NEB. After assessing the quality and quantity of the preparation with UV absorbance, we perform self-cleaving reactions in the presence and absence of 1 μM MgCl2 and 5 μM theophylline and analyzed the products with 7% denaturing polyacrylamide gel electrophoresis (PAGE). We used polyacrylamide gel electrophoresis instead of agarose gel electrophoresis because we need the increased ability of polyacrylamide gels to resolve relatively small differences in RNA size. We used 6 M urea during electrophoresis to establish denaturing conditions that prevent folding of the ribozyme-riboswitch RNA by base pairing. The DNA molecular weight marker used was the Hi-Lo DNA Marker from Bionexus, Inc and the RNA marker used was the Low Range ssRNA Ladder from New England Biolabs, Inc.
RNA Folding Analysis
RNA folding analysis was conducted with ViennaRNA (Vienna RNA, 2005), which uses a traditional computing approach to RNA folding and Qfold, a new approach based on a Quadratic Unconstrained Binary Optimization (QUBO) modeling paradigm (Lewis et al. 2021). Both approach use the canonical Watson-Crick base pairing rules of C paring with G and A pairing with U as well as the non-canonical base pair rule of G pairing with U.
Synthesis of the Ribozyme Chain Molecule
This section describes how we synthesized the ribozyme chain molecule (RCM) shown in Figure 1A. We obtained the Thiol and Glycol DNAs from IDT by ordering 5’ /5ThioMC6-D/AATAGAACTTATGAC 3’ and 5’ /5Phos/ CTACCATGCATGGAC/iSp9/GAATTCGCGGCCGCTTCTAGAGTTGACA 3’, respectively. The thiol medication enables coupling to amino-derivatized MNPs and /iSp9/ is a triethylene glycol spacer. We used T4 RNA ligase from NEB to ligate the 5’ end of the ribozyme-riboswitch RNA to the Thiol DNA and the 3’ end to the Glycol DNA, and analyzed the products with 7% denaturing PAGE. The DNA molecular weight marker used was the Hi-Lo DNA Marker from Bionexus, Inc and the RNA marker used was the Low Range ssRNA Ladder from New England Biolabs, Inc.
Magnetic Nanoparticle Coupling
The MNPs we used were 200 nm Super Mag Amine Beads from Ocean Nanotech. RCMs were prepared for coupling to MNPs by reduction with 50 mM DTT, and purification on a NAP-25 column. We coupled the MNPs to RCMs with sulfosuccinimidyl 4-(N-maleimidomethyl) cyclohexane-1-carboxylate, prepared as a 25 mg/ml solution in DMSO. We used a strong conventional magnet to draw coupled MNP-RCMs to the side of a microcentrifuge tube, removed the remaining liquid, and Resuspend the MNP-RCMs in TE buffer.
Testing Ribozyme-Riboswitch Function
We tested theophylline-dependent ribozyme-riboswitch self-cleavage of RCMs using two different CDM gene constructs with sequences that can be found as J119302 (medium promoter, medium RBS) and J119303 (high promoter, high RBS) in the Registry of Standard Biological Parts (MIT Working Group, 2005). We used GoTaq Green from Promega Corporation for these traditional PCRs using Spacer (5’ GAATTCGCGGCCGCTTCTAGAGTTGACA 3’) as the forward primer and Short Rev 2 (5’ CTGCAGCGGCCGCTAC 3’) as the reverse primer. We conducted 20 cycles of 94℃ for 15 seconds, 55℃ for 15 seconds, and 94℃ for 30 seconds. Products were analyzed with 1% agarose gel electrophoresis using the Hi-Lo DNA Marker from Bionexus, Inc.
Recombinase Polymerization Amplification
We used a kit from TwistDX for Recombinase Polymerization Amplification (RPA) experiments (TwistDX 2005). RPAs were conducted in a 37℃-water bath for 20 minutes with 1 ng of starting template, using Prefix_for (GAATTCGCGGCCGCTTCTAGAG 3’) as the forward primer and Short Rev 2 (5’ CTGCAGCGGCCGCTAC 3’) as the reverse primer. The products were analyzed with 1% agarose gel electrophoresis using the 1 kb Plus DNA Ladder from New England Biolabs, Inc.
Microfluidic Droplet Production
Because we wanted to develop a system for optimizing in vitro metabolic output, we learned how to produce microfluidic droplets that serve as artificial cellular compartments. Droplet formation occurs by the partitioning of a water-based (aqueous) mixture containing all of the components for our optimization system into numerous micrometer-sized droplets as an emulsion in oil. The droplets are formed by polar-nonpolar interactions of fluids and stabilized with the use of biocompatible surfactants that are dissolved in the oil. As seen in Figure 1B, the droplets are formed by using micrometer-scale channels to control the interaction of a stream of the aqueous solution with streams of the oil. We used the OB1 MK3 Microfluidic Flow Control System from Elveflow to make the droplets. The oil (continuous phase) reservoir was filled with droplet stabilization oil from Droplet Genomics (cat. no. DG-DSO-20). Both the dispersive (aqueous) and continuous (oil) channels were set on 600 mbar and droplets were collected in 1.5 ml tubes.
RESULTS

Figure 2. Ribozyme Structure and Function. A. The most stable structure predicted for the ribozyme by ViennaRNA (Vienna RNA, 2005). B. The most stable structure predicted for the ribozyme by Qfold (Lewis et al. 2021). C. Image of a 12% denaturing polyacrylamide gel showing cleavage of the ribozyme. The DNA marker includes 50, 100, 200, 300, 400, and 500 bp fragments. The single stranded RNA ladder includes 50, 80, 150, 300, and 500 nt fragments. The last lane shows ribozyme-riboswitch self-cleavage products dependent on magnesium and theophylline.
As illustrated in Figure 1, the key to our selection strategy is the ribozyme-riboswitch that engages in theophylline-dependent self-cleavage. This enables the magnetic separation of optimal CDM genotypes that have been freed from their MNPs by ribozyme-riboswitch self-cleavage from suboptimal ones that have not. The ribozyme-riboswitch we chose was discovered with a screen in mammalian cells of synthetic ribozyme-riboswitch libraries (Xiang et al. 2019). Figure 2A shows the sequence of the ribozyme-riboswitch and the manner in which it uses intramolecular base pairing to achieve a folded secondary structure known as a hammerhead. The structure in Panel A was determined by ViennaRNA, which uses a traditional computing approach to evaluate the large space of possible structural solutions and report those that are predicted to be the most stable (Vienna RNA, 2005). Panel B shows the results from a new approach to RNA modeling called Qfold that is based on a Quadratic Unconstrained Binary Optimization (QUBO) modeling paradigm that fits naturally with the parameters and constraints required for RNA folding (Lewis et al. 2021). Even though two different approaches to this RNA folding problem were taken, the fact that the two ribozyme-riboswitch structures are only slightly different provides confidence in their accuracy. For each structure, the point of cleavage is indicated, which converts the 76 nt ribozyme-riboswitch into 70 nt and 6 nt products.
We produced the ribozyme-riboswitch as an RNA product of the T7 RNA polymerase-dependent in vitro transcription of a synthetic DNA template. Figure 2B shows the image of a denaturing polyacrylamide gel that we used to analyze the ribozyme-riboswitch. Lane 3 shows a band at 76 nt for the full-length ribozyme-riboswitch. It also shows bands at 70 nt and 6 nt, demonstrating background cleavage of the ribozyme-riboswitch in the absence of theophylline. Because ribozyme-riboswitch cleavage depends on both MgCl2 and theophylline, we incubated it in the presence and absence of each. Lanes 4-6 show both cleaved and uncleaved RNA, but lane 7 shows much less of the uncleaved RNA, as well as an increase in the intensity of the cleavage products. There are extra bands in lane 7 between 70 nt and 76 nt that are likely due to alternative folding structures of the 70 nt product that were allowed to form because of incomplete denaturation. This result encouraged us to proceed with construction of the RCM.

Figure 3. Construction and Testing of the Ribozyme Chain Molecule (RCM). A. 12% denaturing polyacrylamide gel showing successful ligations of mammal ribozyme to thiol and spacer DNA to produce the RCM primer. The DNA marker includes 50, 100, 200, 300, 400, and 500 bp fragments. The single stranded RNA ladder includes 50, 80, 150, 300, and 500 nt fragments. B. Incubation of RCMs with or without 5 uM theophylline followed by PCR. The DNA marker includes bright bands at 500, 1000, and 3000 bp.
Our first step in building the RCM was to build an amplification primer from the ribozyme-riboswitch and two synthetic DNAs. We used T4 RNA ligase to connect the 5’ end of the ribozyme-riboswitch to the 3’ end of the Thiol DNA linker and the 3’ end to the Glycol DNA spacer. The denaturing polyacrylamide gel image in Figure 3A shows the construction steps. Lanes 3 and 4 contain the 15 nt Thiol DNA and the 43 nt spacer DNA, respectively. Lanes 5-8 show the results of reactions with T4 RNA ligase using an increasing number of RCM components. Although the full product of 76 nt is visible in lane 8, there are several smaller bands in all of the lanes with ligation products. Because there is both a 5’ and a 3’ end available for ligation on both the ribozyme-riboswitch and the spacer DNA, and an available 3’ end on the Thiol DNA, there are several ways in which the fragments can be ligated to produce bands smaller than 76 nt. The band in lane 8 that is just smaller than 76 nt could be a ligation product that includes the 70 nt self-cleaved riboswitch-ribozyme RNA.
Our next step was to couple the RCM primer to MNPs using the thiol group on the 5’ end of the RCM and the amino derivative on the MNPs. We tested the resulting product by using it in standard PCR with a DNA reverse primer and either of two CMD gene expression units as templates. We incubated the PCR products in the presence and absence of ribozyme-riboswitch self-cleavage conditions, which include both MgCl2 and theophylline, and separated the MNPs with a magnet before conducting PCR again to quantitate ribozyme-riboswitch self-cleavage. If self-cleavage of the ribozyme-ribozyme occurs, the RCM cannot serve as a primer for PCR of the CDM units. If the RCM remains intact, it can be used during PCR to produce a product of 3346 bp. This product is highlighted by the red box in Figure 3B. There is more of the product in lane 3 than in lane 2, and much more in lane 5 than in lane 4, indicating that ribozyme-riboswitch self-cleavage occurred more often in the absence of theophylline than in its presence. Combined with the results of Figure 2C, this indicates that the behavior of the ribozyme-riboswitch was reversed when it was coupled to MNPs. We consider this unexpected result further in the Discussion section. Figure 3B also shows extra bands that are likely due to nonspecific annealing of primers during PCR.

Figure 4. Recombinase Polymerase Amplification (RPA). Image of a 1% agarose gel showing successful Recombinase Polymerase Amplification (RPA) using a kit from TwistDX (TwistDX 2005). The DNA marker includes 50, 100, 200, 300, 400, 500, 750, 1400, 1550, 2000, 3000, 4000, and 6000 bp fragments.
Our plan is to use the ribozyme-included and MNP-coupled forward primer inside a given microfluidic droplet to amplify a single caffeine demethylase gene expression cassette genotype. Because there are enzymes and other temperature-sensitive molecules in the microfluidic droplets, we cannot use the extreme temperature (94°C) needed for the denaturation step in standard PCR. Recombinase Polymerase Amplification (RPA) is an isothermal DNA amplification method that achieves denaturation with the strand invasion activity of a recombinase (Lobato and O’Sullivan 2018). The results of our experiments to optimize RPA are shown in Figure 4. The main technical barrier to the use of RPA for our project is the amplification of the CDM gene expression cassette, which is 3274 bp. Although TwistDX, the manufacturer of the RPA kit we used, advertises that RPA has been used to amplify at most 500 bp, we were able to improve upon that (TwistDX 2005). Figure 4 shows that we were able to use RPA to amplify a series of increasingly larger templates. Although the longest of these is 1186 bp, it is still short of our goal of amplifying 3000 bp. We consider this technical limitation further in the Discussion section.
DISCUSSION
Conclusions
Although successful implementation of our RCM selection strategy is a considerable biological engineering challenge, we made some significant progress toward it. We were able to build a primer composed of a Thiol DNA linker, a ribozyme, and a Glycol DNA spacer, to couple the primer to an MNP, and to use it for PCR amplification of a gene expression cassette. We measured theophylline-dependent behavior of a ribozyme-riboswitch alone and in the context of an RCM. We were able to make microfluidic aqueous droplets in an oil suspension, include MNPs in the droplets, and use a magnet to separate them. We learned how to conduct isothermal DNA amplification using RPA in support of our plan to amplify a single gene expression genotype in each microfluidic droplet.
Technical Limitations and Sources of Error
Although our experimental results provide preliminary support for the feasibility of using a self-cleaving ribozyme-riboswitch as the basis of a new selection strategy for metabolic engineering, we encountered several technical barriers to a complete demonstration of it. We were encouraged by our ability to optimize RPA and increase product size from about 500 bp to almost 1500 bp, but a technical barrier to the successful demonstration of our RCM strategy is our inability to use RPA to amplify full-length CDM gene expression units, which are more than 3000 bp. Variables in the standard RPA protocol that could be explored to increase product size include the reaction temperature, which affects the amount of thermal energy available for DNA denaturation, the concentration of magnesium, which impacts DNA denaturation via shielding of phosphates by magnesium cations, and the ratio of primer to template DNA, which affects the ability of recombinase to facilitate primer binding. Alternatively, we could transition to the use of a smaller gene expression cassette, although this would require finding a new metabolic reaction to optimize and a ribozyme-riboswitch that can transduce its output.
Another technical barrier to successful implementation of our strategy for optimization of metabolic pathways relates to our growing understanding of the nature of highly concentrated solutions of small and large molecules that occur inside cells. There is a growing appreciation that density-dependent phase separation in cells leads to liquid-like non-Newtonian biomolecular condensates that play both structural and metabolic roles and that metabolites such as ATP serve as hydrotropes to maintain the solubility of proteins (Banani et al. 2017). This increasingly sophisticated view of a saturated solution of molecules in the cytoplasm of cells might also apply to aqueous microfluidic droplets. If density- and temperature-dependent phase separation happens in droplets, it could dramatically affect both gene expression and metabolic output. In fact, we previously observed a large dynamic range of GFP output from droplets that were made with a gene expression cassette and an E. coli cellular extract (Koucky et al. 2021). Although the effects of phase separation could confound efforts to use selection to optimize metabolic output, a better understanding of it might lead to ways that it could be controlled.
Our hypothesis that the theophylline dependence of the ribozyme-riboswitch will be retained in the context of an RCM coupled to an MNP was not supported by our results. The theophylline dependence of ribozyme-riboswitch self-cleavage was reversed when the ribozyme-riboswitch was incorporated into the RCM. Although this means that our selection strategy will not work as originally designed, we can conceive of a reversed strategy in which optimal genotypes coupled to MNPs are attracted to a magnet because their ribozyme-riboswitches are prevented from self-cleaving by theophylline. Our observation also has implications for the understanding of ribozyme-riboswitch function. In the context of an RNA molecule such as an mRNA transcript, intramolecular base pairing drives the aptamer of the theophylline ribozyme-riboswitch to fold in a manner that prevents ribozyme self-cleavage. Noncovalent interactions with theophylline alter the pattern of base pairing to generate a three-dimensional structure that supports this reaction. According to our evidence, the reaction is affected by having the ends of the ribozyme-riboswitch RNA constrained differently in the RCM than they are in an RNA molecule. The effect of the MNP on altering the process by which theophylline alters RNA folding might be especially large.
There are potential sources of error in our experiments that should be pointed out to place our results in the proper context. Because we did not clone and sequence the riboswitch-ribozyme, we must assume that in vitro transcription faithfully reproduced the correct RNA sequence from the synthetic DNA template. If changes in the sequence were introduced, then we produced a population of riboswitch-ribozyme sequences instead of a single sequence. This would affect each of our downstream ribozyme-riboswitch experiments. Another source of error comes from the background self-cleaving activity of the ribozyme-riboswitch. Both the 70 nt and the 6 nt cleavage products can participate in the T4 RNA ligase reactions for RCM construction, which means that we produced not only the full-length RCM, but a variety of other products that cannot function as RCMs. Inefficient magnetic separation of the RCMs, background self-cleavage in the absence of theophylline, and nonspecific primer annealing are potential sources of error in the experiment in which we subjected RCMs to self-cleavage conditions before we used them in standard PCR.
Significance and Implications
The significance of our study derives from its implications for the advancement of metabolic engineering. Our idea of transducing metabolic output into fitness by using a riboswitch-ribozyme that binds to the product of the reaction by which caffeine demethylase converts caffeine to theophylline could be generalized to other single or multistep anabolic or catabolic reactions, as long as riboswitch-ribozymes with appropriate specificities are available. Our proposal to carry out selection in microfluidic droplets enables the simultaneous evaluation of many possible genotypes and allows for the inclusion of metabolites that are toxic to cells. Our observation of reversed riboswitch-ribozyme behavior in the MNP context compared to the RNA context has implications for metabolic engineering in terms of how we understand the science of ribozyme-riboswitch function and how we exploit that knowledge for metabolic engineering. Our overall approach to the use of selection for optimization of metabolism could lead to metabolic engineering applications in a variety of areas, including biosynthesis of pharmaceuticals, production of biofuels, chemical commodity synthesis, and bioremediation.
ACKNOWLEDGEMENTS
Support from National Science Foundation (http://www.nsf.gov/) RUI grant MCB-1613281 to Missouri Western State University is gratefully acknowledged. We would also like to thank Dr. Mark Lewis and Dr. Amit Verma for providing the RNA folding images.
REFERENCES
Bali, A.P. (2019) ‘Selection-aided Metabolic Engineering of Microbial Cell Factories for Vitamin Production in Escherichia coli’, available: https://orbit.dtu.dk/en/publications/selection-aided-metabolic-engineering-of-microbial-cell-factories [accessed 21 Dec 2020].
Banani, S.F., Lee, H.O., Hyman, A.A., Rosen, M.K. (2017) ‘Biomolecular condensates: organizers of cellular biochemistry’, Nature Reviews Molecular Cell Biology, 18(5), 285–298.
Bawa, A.S., Anilakumar, K.R. (2013) ‘Genetically modified foods: safety, risks and public concerns—a review’, Journal of Food Science and Technology, 50(6), 1035–1046.
Breaker, R.R. (2012) ‘Riboswitches and the RNA World’, Cold Spring Harbor Perspectives in Biology, 4(2), a003566.
Choe, J.H., Williams, J.Z., Lim, W.A. (2020) ‘Engineering T Cells to Treat Cancer: The Convergence of Immuno-Oncology and Synthetic Biology’, Annual Review of Cancer Biology, 4(1), 121–139.
Doudna, J.A., Cech, T.R. (2002) ‘The chemical repertoire of natural ribozymes’, Nature, 418(6894), 222–228.
Eckdahl, T.T., Campbell, A.M., Heyer, L.J., Poet, J.L., Blauch, D.N., Snyder, N.L., Atchley, D.T., Baker, E.J., Brown, M., Brunner, E.C., Callen, S.A., Campbell, J.S., Carr, C.J., Carr, D.R., Chadinha, S.A., Chester, G.I., Chester, J., Clarkson, B.R., Cochran, K.E., Doherty, S.E., Doyle, C., Dwyer, S., Edlin, L.M., Evans, R.A., Fluharty, T., Frederick, J., Galeota-Sprung, J., Gammon, B.L., Grieshaber, B., Gronniger, J., Gutteridge, K., Henningsen, J., Isom, B., Itell, H.L., Keffeler, E.C., Lantz, A.J., Lim, J.N., McGuire, E.P., Moore, A.K., Morton, J., Nakano, M., Pearson, S.A., Perkins, V., Parrish, P., Pierson, C.E., Polpityaarachchige, S., Quaney, M.J., Slattery, A., Smith, K.E., Spell, J., Spencer, M., Taye, T., Trueblood, K., Vrana, C.J., Whitesides, E.T. (2015) ‘Programmed Evolution for Optimization of Orthogonal Metabolic Output in Bacteria’, PLOS ONE, 10(2), e0118322.
Flores Bueso, Y., Lehouritis, P., Tangney, M. (2018) ‘In situ biomolecule production by bacteria; a synthetic biology approach to medicine’, Journal of Controlled Release, 275, 217–228.
Hughes, R.A., Ellington, A.D. (2017) ‘Synthetic DNA Synthesis and Assembly: Putting the Synthetic in Synthetic Biology’, Cold Spring Harbor Perspectives in Biology, 9(1), a023812.
Jaiswal, S., Shukla, P. (2020) ‘Alternative Strategies for Microbial Remediation of Pollutants via Synthetic Biology’, Frontiers in Microbiology, 11, available: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7249858/ [accessed 21 Dec 2020].
Koucky, O., Wagner, J., Aguilera, S., Bashaw, B., Chen, Q., Eckdahl, A., Edman. E., Gomez, P., Hanlan, N., Kempf, N., Mattoon, D., McKlin, S., Mazariegos, C., Morehead, A., Ong, S.Q., Peterson, A., Rojas, M., Roland, K., Schildknecht, K., Seligmann, H., Slater, K., Tauchen, A., Tittor, R., Travieso, T., Urban, D., Willis, C., Zhou, J., Snyder, N.L., Heyer, L.J., Poet, J.L., Eckdahl, T.T., Campbell, A.M. (2021) ‘Synthetic Biology Bicistronic Designs Support Gene Expression Equally Well in vitro and in vivo’, American Journal of Undergradute Research, 17(1).
Kumar, A. (2020) ‘Synthetic Biology and Future Production of Biofuels and High–Value Products’, in Kumar, A., Yau, Y.-Y., Ogita, S. and Scheibe, R., eds., Climate Change, Photosynthesis and Advanced Biofuels: The Role of Biotechnology in the Production of Value-Added Plant Bio-Products, Springer: Singapore, 271–302, available: https://doi.org/10.1007/978-981-15-5228-1_11 [accessed 21 Dec 2020].
Lewis, M., Verma, A., Eckdahl, T.T. (2021) ‘Qfold: A new Modeling paradigm for the RNA folding problem’, Journal of Heuristics, In Press.
Lobato, I.M., O’Sullivan, C.K. (2018) ‘Recombinase polymerase amplification: Basics, applications and recent advances’, TrAC Trends in Analytical Chemistry, 98, 19–35.
Lu, Y. (2017) ‘Cell-free synthetic biology: Engineering in an open world’, Synthetic and Systems Biotechnology, A tribute to Arny Demain, for his lifelong pioneering contributions to biochemical engineering, 2(1), 23–27.
Meng, F., Ellis, T. (2020) ‘The second decade of synthetic biology: 2010–2020’, Nature Communications, 11(1), 5174.
MIT Working Group. (2005). Registry of Standard Biological Parts. Retrieved December 18, 2020, from http://partsregistry.org/Main_Page
Ojuederie, O.B., Babalola, O.O. (2017) ‘Microbial and Plant-Assisted Bioremediation of Heavy Metal Polluted Environments: A Review’, International Journal of Environmental Research and Public Health, 14(12), available: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5750922/ [accessed 21 Dec 2020].
Parts.Igem.Org [online] (2020) available: http://parts.igem.org/Main_Page [accessed 21 Dec 2020].
Pontrelli, S., Fricke, R.C.B., Sakurai, S.S.M., Putri, S.P., Fitz-Gibbon, S., Chung, M., Wu, H.-Y., Chen, Y.-J., Pellegrini, M., Fukusaki, E., Liao, J.C. (2018) ‘Directed strain evolution restructures metabolism for 1-butanol production in minimal media’, Metabolic Engineering, 49, 153–163.
Quianzon, C.C., Cheikh, I. (2012) ‘History of insulin’, Journal of Community Hospital Internal Medicine Perspectives, 2(2), available: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3714061/ [accessed 21 Dec 2020].
Sherlock, M.E., Sudarsan, N., Stav, S., Breaker, R.R. (2018) ‘Tandem riboswitches form a natural Boolean logic gate to control purine metabolism in bacteria’, eLife, 7, e33908.
TwistDX. (2005). RPA. The Versitile PCR Replacement. Retrieved December 18, 2020, from https://www.twistdx.co.uk/
Vienna RNA. (2005). Vienna RNA Web Services. Retrieved December 4, 2020, from http://rna.tbi.univie.ac.at/
Xiang, J.S., Kaplan, M., Dykstra, P., Hinks, M., McKeague, M., Smolke, C.D. (2019) ‘Massively parallel RNA device engineering in mammalian cells with RNA-Seq’, Nature Communications, 10(1), 4327.