

Michael A. Lu and Tina Gibson
The Mississippi School for Mathematics and Science, 1100 College St, Columbus, MS 39701
ABSTRACT
Cancer is a leading cause of high mortality rates around the world. Many scientists have explored anticancer peptides (ACPs), which are peptides with anti-tumor activity that can be safer than conventional drugs due to high activity coupled with high selectivity and delivery control. However, current in vitro methods of discovery are both time-consuming and expensive. This study aims to use modern machine learning tools to discover new ACP candidates. Discriminative models were trained to classify between anti-cancer and non-anti-cancer peptides using data from several anti-cancer peptide databases, totaling 584 known ACPs. The best-performing model, the support vector machine (SVM) could identify with a 90.4 percent whether a given peptide carried anticancer properties. A long short-term memory (LSTM) recurrent network was also developed as a method of ACP generation. The model created 40 preliminary sequences, and the sequences were verified through various computational models and literature-based properties of ACP’s, concluding that around 90 percent of the generated sequences carry anticancer properties. This work illustrates the potential of generative modeling techniques to facilitate the discovery of new anticancer peptides.
INTRODUCTION
Cancer-related diseases continue to affect millions of individuals around the world every year, and thus, it is important to explore new alternative treatments. Drugs are used for cancer treatments, and although they are beneficial, they can also cause harmful side effects. Anticancer peptides (ACPs) have the potential to be a form of alternative treatment that avoids those harmful side effects. In fact, they have many comparative advantages over conventional cancer drugs such as higher levels of activity, more specificity and affinity, and they are less immunogenic and have better delivery control (Gholibeikian et al., 2019). ACPs typically range from 10-30 residues in length. There are three main types of ACPs: pore-forming peptides that target the cell membrane of cancer cells; cell penetration peptides that enter the cell, and tumor-targeting peptides that bind to the receptors on cancer cells (Marqus et al., 2017).
Currently, there are only ten anticancer peptides that are currently being developed as drugs (Shoombuatong et al., 2018). More research is needed in this field, as explained by Yin et al. (2019). Furthermore, current in vitro methods of discovery are both time-consuming and expensive (Manavalan et al., 2017), demonstrating an unmet need for more effective ways to create anticancer peptide candidates before proceeding into in vitro testing. Machine learning has the potential to fill this role. Given enough data, machine learning models can learn patterns in ACPs and have the potential to generate new peptides from them. This study aims to use machine learning models to generate new candidates to be synthesized. Additionally, because different machine learning classifier models are better for tasks with varying levels of complexity, a goal of this study is to test a wide array of common machine learning predictor models to predict ACPs.
Many machine learning applications in this field are focused on creating predictors that classify whether given peptides are ACPs. These classifiers are able to predict peptides successively, but they lack the ability to generate new ACP candidates. Generative machine learning is a type of machine learning that analyzes existing datasets to generate new instances of data. One type of generative model is the long short-term memory recurrent neural network, which generates new sequences of data. This type of model has been used in the successful generation of antimicrobial peptides (Youmans, 2019). For this reason, an LSTM recurrent neural network can likely be applied to anticancer peptides. A second goal of this study was to test the ability for LSTM recurrent neural networks to construct new anticancer peptide candidates.
In this study, there are two central aims. The first is to train a variety of different predictive models to determine when a random peptide is an ACP, and the second is to create a generative model to predict new ACP candidates. To achieve these two goals, machine learning classifiers and generative models can be trained, and with further development, these tools can help facilitate the discovery of new ACP’s as an alternative treatment to cancer
METHODS
Classification Models
Classification models of varying model complexity were explored to determine the optimal model type for classifying ACP and non-ACPs. The four following classification models were used and are described in order of model complexity. The first model is the K-nearest neighbor (KNN) classifier, which predicts a class based on the similarity of the features of a sample to its neighbors (Peterson et al., 2009). The second model is Support Vector Machine, a model that finds the hyperplane which maximizes the margins between two classes (Suykens et al., 1999). The third model is Random Forest (RF), which is an ensemble learning method that utilizes a group of weak predictors known as decision trees (Liaw et al., 2002). The fourth model is a multi-layer perceptron (MLP), which is a feedforward artificial neural network containing multiple layers of artificial neurons that use a nonlinear activation function to propagate information from input to output (Pal et al., 1992).
Generative Models
To generate new and realistic sequences of anti-cancer peptides, a long short-term memory model (Hochreiter and Schmidhuber, 1997), which is a type of recurrent neural network, was used to generate new sequences from a training set of established ACPs. LSTM is composed of a cell, an input gate, an output gate, and a forget gate, and it is used to retain long-term information in sequential data to use in conjunction with short term data to make predictions. The generative models use nonlinear activation functions such as hyperbolic tangent or sigmoid functions to allow the model to “remember” or “forget” the previous input data in the sequence. The output of the LSTM layer is fed to linear layers, a dropout layer, and a softmax activation layer. Linear layers contain a series of interconnected nodes and nonlinear activation functions to transform the data. Dropout layers allow the model to adaptively select relevant features to include. The softmax layer allows the model to transform the output to a probability value for each possible amino acid class. The amino acid class that has the highest predicted probability is determined to be the most probable amino acid to follow. This process is repeated by shifting the prediction one unit to the right until the model has predicted the full peptide sequence.
The LSTM models generate new anticancer peptides by sampling the first L amino acids of known ACP’s where L is the size of the window the model was trained on. In addition to possible amino acid classes, an additional “sequence end” class was added, which allows the model to learn to terminate sequence generation. During prediction, the LSTM model generates each amino acid until the “sequence end” class is predicted.
Data Collection and Processing
Datasets of verified anticancer peptides were collected from three databases. The first is the Data Repository of Antimicrobial Peptides (DRAMP) database (Fan et al., 2016; Liu et al., 2017; Liu et al., 2018; Kang et al., 2019), consisting of 74 tested ACPs. The second is the Antimicrobial Peptide Database (APD) (Wang and Wang, 2004; Wang et al., 2016; Wang et al., 2009), which includes 219 verified ACPs. The third is the Anticancer Peptide and Protein Database (CancerPPD) of 374 experimentally verified ACPs. Sequences shorter than 12 residues were removed, and the datasets were combined.
For the non-ACP class, 584 unique anticancer peptide sequences were sampled randomly from the Swiss Protein database (UniProt Consortium, 2019) which contains 561,568 annotated and reviewed peptide sequences. These naturally-occurring sequences were between the lengths of 10-25 residues. 11 categories of physicochemical features were extracted from each peptide sequence with the PydPi (Drug-Protein Interaction with Python) library (Cao et al., 2013). These 11 categories of features contain 2049 total features (e.g., amino acid composition, charge, hydrophobicity, etc.), and were normalized in the range from 0 to 1 for each feature as follows:

Then, two feature selection methods were tested. The chi-squared test (Forman et al., 2003) was tested to find the features with the least variance for each class. The equation is given by:

To use the generative model for realistic sequence generation, several data-processing steps were performed. A sliding window technique was used to generate training and test datasets. For example, the first eight amino acids in a sequence can be used to predict the following amino acid, and this window is shifted to the right to create additional samples.

Machine learning models require numerical data as input; therefore, peptide sequences have to be transformed from an alphabet representation to a numerical representation. We used one-hot encoding, which expresses each amino acid as a vector the size of the alphabet, with the location of the number 1 identifying the letter. For example, if there are three letters in the alphabet [“a”, “b”, “c”], then the sequence “abca” would be represented as [1, 0, 0], [0, 1, 0], [0, 0, 1], [1, 0, 0]. After encoding the sequences into one-hot vectors, the input is then formatted for the model. LSTM models use a 3-dimensional array as inputs, so the input data were reshaped to be (Batch Size, Time Steps, Features). Batch size represents the number of data samples created from the shifting window, time steps refer to the size of the shifting window, and features are the one-hot encoded data.
Metrics for evaluation
Many metrics were used to validate the effectiveness of the models. A simple accuracy metric was used to find the percentage of test and training samples the model could identify, specificity and sensitivity were found, and the F1 score was also calculated from a combination of precision and recall to balance the influence of false positives and false negatives. These four metrics can be defined using true positives (TP), true negatives (TN), false positives (FP), and false negatives (FN).
The following metrics are used:

RESULTS
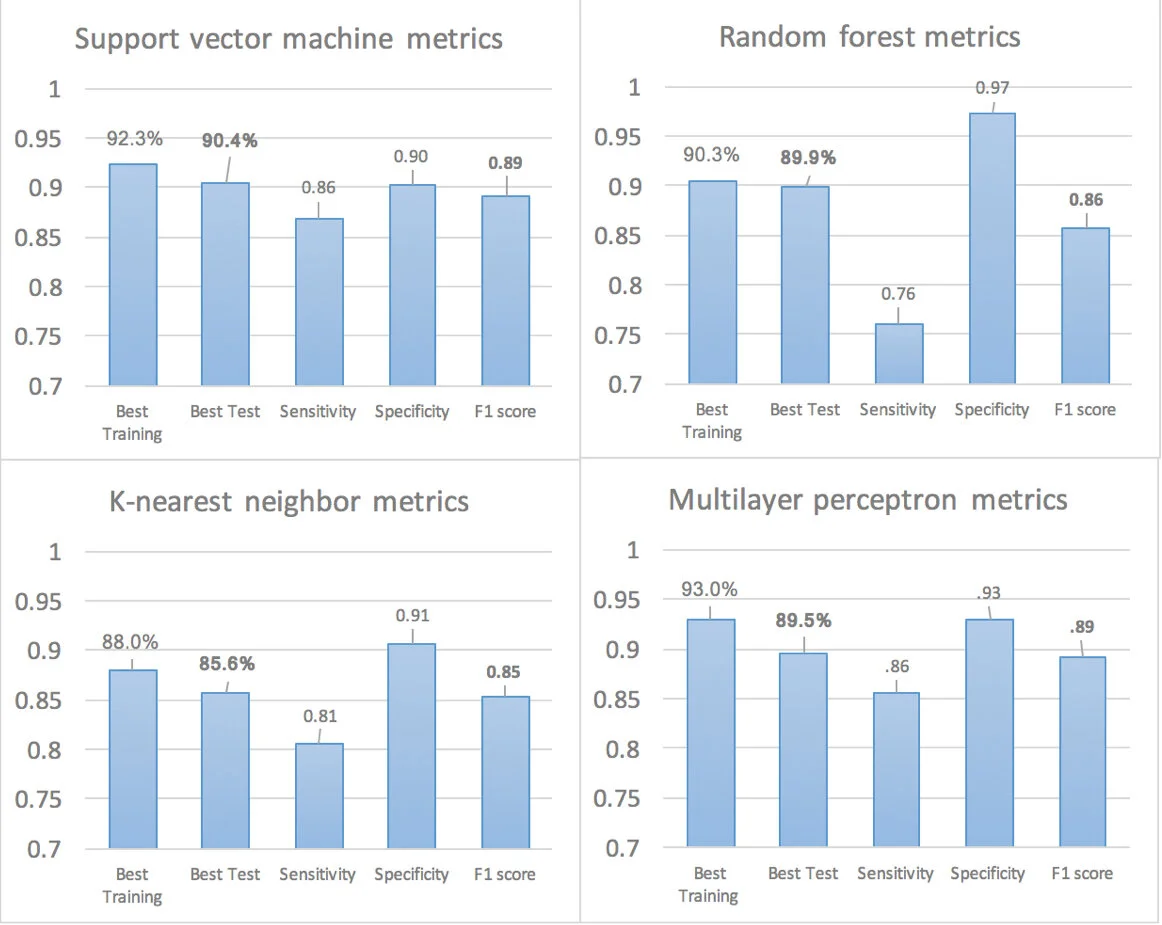
Figure 1. Metrics for predictor models. After the models were trained, each model was tested 25 times against a test dataset. The top-performing model’s training accuracy, testing accuracy, sensitivity, specificity, and F1-score is recordedsensitivity, specificity, and F1-score is recorded.
Predictor model final evaluation
To verify the different types of models, the initial dataset was split to 60 percent training data and 40 percent testing data. After the models are optimized for their parameters and trained, the average training and test accuracies are recorded over 25 iterations. Then, the top-performing model’s training accuracy, testing accuracy, sensitivity, specificity, and F1-score are recorded. The results are shown in Figure 1.
SVM: The support vector machine achieved the highest accuracy over the other tested models. It achieved an average training and testing accuracy of 93.4 percent and 89.1 percent respectively, and the top-performing model scored a 92.3 percent training accuracy, a 90.4 percent testing accuracy, a sensitivity of 0.87, a specificity of 0.90, and an F1-score of 0.89.
Random forest: The random forest model achieved an average training and testing accuracy of 90.3 percent and 86.7 percent respectively, and the top-performing model scored a 90.4 percent training accuracy, an 89.9 percent testing accuracy, a sensitivity of 0.76, a specificity of 0.97, and an F1-score of 0.86.
K-nearest neighbors: The k-nearest neighbors algorithm had a training and testing accuracy of 88.0 percent and 85.7 percent respectively, and it scored a sensitivity of 0.81, a specificity of 0.91, and an F1-score of 0.85.
Multilayer perceptron: The multilayer perceptron model achieved an average training and testing accuracy of 92.5 percent and 88.9 percent respectively, and the top-performing model scored a 93.0 percent training accuracy, an 89.5% testing accuracy, a sensitivity of 0.85, a specificity of 0.93, and an F1-score of 0.89.

Figure 2. The initial SVM predictor’s probabilities for the 40 generated sequences with a line signifying the 90% confidence threshold.
Verifying generated peptides

Figure 3. The ACPPred, ACPPred-FL, and Tyagi web server predictors’ probabilities for the 40 generated sequences with a line signifying the 90% confidence threshold.
First, the 40 generated peptides were tested against the top-performing SVM model, and the predicted probabilities are shown in Figure 2. Then, the 40 generated were tested against the Anticancer Peptide Predictor (ACPPred) (Schaduangrat et al., 2019), Anti-Cancer peptide Predictor with Feature representation Learning (ACPPred-FL) (Wei et al., 2019), and Tyagi (Tyagi et al., 2013) webserver predictors. The predicted probabilities are shown in Figure 3.

The 40 peptides were then tested against six literature-based indicators of anticancer peptides shown in Table 1. First of all, anticancer peptides are known to exhibit a net positive charge and a high hydrophobicity (Shoombuatong et al., 2018). The forty preliminary sequences were calculated to have a +5.08 positive charge and a 41.30 hydrophobicity value. The optimal length of an anticancer peptide is found to be between the lengths of 21-30 residues long, and the average length of the forty sequences is 22.22 residues long. The three most prevalent amino acids in anticancer peptides are Glycine (10.88 percent), Lysine (10.25 percent), and Leucine (11.23 percent) (Shoombuatong et al., 2018). These amino acids are also seen to be common amino acids in the generated sequences. Lysine (21.73 percent) and Leucine (12.95 percent) are the most predominant with Glycine (7.54 percent) as the fifth most common. Then, ACP’s typically begin with a Glycine, Leucine, Alanine, and Phenylalanine, and 60 percent of the generated ACP candidates follow this pattern. Furthermore, ACP’s usually end with a Valine, Cysteine, Leucine, and Lysine, and 53 percent of the generated ACP candidates follow this pattern. The presence of these amino acids at the beginning and ending of the peptides may be important for the peptides to penetrate through tumor cell membranes. The 40 generated ACP drug candidates exhibit many known properties of anticancer peptides.
DISCUSSION
Four types of machine learning models were used to create predictors of whether a peptide had anticancer peptides. The significance of the predictive models was found by creating a test dataset and calculating metrics like accuracy and F1 score. The top-performing SVM model achieved a 90.4 percent testing accuracy and an F1 score of 0.89, which is comparable to other published predictive models (Tyagi et. al., 2013; Manavalan et al., 2017; Schaduangrat et al., 2019). Given a random peptide, the models will be able to identify whether that peptide is an ACP with a 90 percent accuracy. Although the model can predict whether a peptide is an ACP with relatively high accuracy, a potential limitation of the model is its inability to identify which type of cancer the peptides target. It is also important to validate the models by testing the peptides experimentally. To improve the model in the future, training on datasets that sort ACP’s by cancer type would allow for cancer-specific peptides to be generated. In addition, training data availability can cause generalization error and is a limiting factor for model prediction performance. As the ACP databases expand, more training data would be available to improve the generalizability of the machine learning models.
These predictive models can contribute to future cancer research in a few ways. First of all, these models can be used to predict favorable mutations for known ACPs to design an ACP containing each favourable change. Second of all, these models can be used to predict new ACPs found in nature. Because ACPs have been found in nature in the past, then it is likely that many peptides in nature have undiscovered anticancer properties. The TrEMBL dataset (UniProt Consortium, 2019) includes over 177 million unreviewed peptides found in nature, and these predictors can make preliminary predictions for ACP candidates.
Furthermore, this study generated new anticancer peptides using machine learning. Current literature is focused on predicting whether a peptide is an ACP. In this project, a long short term recurrent neural network was trained on 584 known anticancer peptides to generate 40 new anticancer peptide candidates. These candidates were verified through self-created predictors, web server predictors, and known properties of ACPs, which have enhanced the accuracy of the models. Furthermore, these peptides were tested with other published models for ACP’s in the field (Figure 3), showing that a majority of the peptides fit to other scientists’ models (Tyagi et al., 2013; Schaduangrat et al., 2019; Wei et al., 2019). In addition, the properties of these peptides follow with the interpretations and knowledge of known anticancer peptides (Table 1 and section 3.2). For instance, a majority of this study’s generated candidates exhibit a positive charge. However, biological functions of the traits remain to be investigated.
The overall application of these predictive and generative models is to create new candidates for ACPs before chemical synthesis and biological screening. The typical drug discovery process of scanning through thousands of libraries of chemical compounds is both time consuming and expensive, so these models can be used to preselect a number of likely candidates before going into testing. In future studies, to verify the models in a lab setting, the peptides generated in this study can be synthesized and tested on various cancer lines to determine their actual efficacy. Analyses using these models to create candidates can increase the likelihood of success for the development of pharmaceutical drugs. With further development, these models can bring potential positive impacts on the discovery process for cancer treatments.
ACKNOWLEDGMENTS
The authors thank Dr. Shien Lu for critical review of the manuscripts.
REFERENCES
Cao, D. S., Liang, Y. Z., Yan, J., Tan, G. S., Xu, Q. S., and Liu, S. (2013). PyDPI: freely available python package for chemoinformatics, Bioinformatics, and chemogenomics studies (accessed in December 2019).
Fan, L., Sun, J., Zhou, M., Zhou, J., Lao, X., Zheng, H. and Xu, H. (2016). DRAMP: a comprehensive data repository of antimicrobial peptides, Scientific Reports, 6,24482. https://doi.org/10.1038/srep24482
Forman, G. (2003). An extensive empirical study of feature selection metrics for text classification, Journal of Machine Learning Research, 3, 1289-1305.
Gholibeikian, M., Bamoniri, A., HoushdarTehrani, M. H., Mirjalili, B. B. F., and Bijanzadeh, H. R. (2019). Structure-activity relationship studies of Longicalcynin A analogues, as anticancer cyclopeptides, Chemico-biological interactions, 108902. doi: 10.1016/j.cbi.2019.108791.
Goldberg, Y., and Levy, O. (2014). word2vec Explained: deriving Mikolov et al.’s negative-sampling word-embedding method. arXiv preprint arXiv:1402.3722. (accessed in December 2019).
Granitto, P. M., Furlanello, C., Biasioli, F., and Gasperi, F. (2006). Recursive feature elimination with random forest for PTR-MS analysis of agroindustrial products, Chemometrics and Intelligent Laboratory Systems, 83(2), 83-90. doi: 10.1016/j.chemolab.2006.01.007.
Hochreiter, S. and Schmidhuber, J. (1997). Long short-term memory. Neural computation, 9(8), 1735-1780.
Kang, X., Dong, F., Shi, C., Liu, S., Sun, J., Chen, J., Li, H., Xu, H., Lao, X., and Zheng, H. (2019). DRAMP 2.0, an updated data repository of antimicrobial peptides, Scientific Data, 6(1), 148. doi: 10.1038/s41597-019-0154-y.
Liaw, A., and Wiener, M. (2002). Classification and regression by random forest, R news, 2(3), 18-22.
Liu, S., Bao, J., Lao, X. and Zheng, H (2018). Novel 3D Structure Based Model for Activity Prediction and Design of Antimicrobial Peptides, Scientific Reports, 8(1):11189. doi: 10.1038/s41598-018-29566-5.Liu, S., Fan, L., Sun, J., Lao, X. and Zheng, H. (2017). Computational resources and tools for antimicrobial peptides, Journal of peptide science : an official publication of the European Peptide Society,23(1):4-12. https://doi.org/10.1002/psc.2947.
Manavalan, B., Basith, S., Shin, T. H., Choi, S., Kim, M. O. and Lee, G. (2017). MLACP: machine-learning-based prediction of anticancer peptides, Oncotarget, 8(44). doi: 10.18632/Oncotarget.20365
Marqus, S., Pirogova, E. and Piva, T. J. (2017). Evaluation of the use of therapeutic peptides for cancer treatment, Journal of Biomedical Science, 24(1), 21. https://doi.org/10.1186/s12929-017-0328-x.
Pal, S. K., and Mitra, S. (1992). Multilayer perceptron, fuzzy sets, classification. (accessed in November 2019).
Peterson, L. E. (2009). K-nearest neighbor. Scholarpedia, 4(2), 1883. (accessed in November 2019).
Schaduangrat, N., Nantasenamat, C., Prachayasittikul, V. and Shoombuatong, W. (2019). ACPred: a computational tool for the prediction and analysis of anticancer peptides, Molecules, 24(10), 1973. https://doi.org/10.3390/Molecules24101973.
Shoombuatong, W., Schaduangrat, N. and Nantasenamat, C. (2018). Unraveling the bioactivity of anticancer peptides as deduced from machine learning, EXCLI journal, 17, 734. doi: 10.17179/excli2018-1447.
Suykens, J. A. and Vandewalle, J. (1999). Least squares support vector machine classifiers, Neural processing letters, 9(3), 293-300. https://doi.org/10.1023/A:1018628609742.
Tyagi, A., Kapoor, P., Kumar, R., Chaudhary, K., Gautam, A. and Raghava, G. (2013). In Silico Models for Designing and Discovering Novel Anticancer Peptides. Scientific Reports, 3(1). https://doi.org/10.1038/srep02984.
UniProt Consortium. (2019). UniProt: a worldwide hub of protein knowledge. Nucleic Acids Research, 47(D1), D506-D515.
Wang, G., Li, X. and Wang, Z. (2016). APD3: the antimicrobial peptide database as a tool for research and education, Nucleic Acids Research, 44, D1087-D1093. doi: 10.1093/nar/gkv1278.
Wang, G., Li, X. and Wang, Z. (2009). APD2: the updated antimicrobial peptide database and its application in peptide design, Nucleic Acids Research, 37, D933-D937. doi: 10.1093/nar/gkn823.
Wang, Z. and Wang, G. (2004). APD: the antimicrobial peptide database, Nucleic Acids Research, 32, D590-D592. https://doi.org/10.1093/nar/gkh025.
Wei, L., Zhou, C., Chen, H., Song, J. and Su, R. (2018). ACPred-FL: a sequence-based predictor using effective feature representation to improve the prediction of anti-cancer peptides, Bioinformatics, 34(23), 4007-4016. doi: 10.1093/Bioinformatics/bty451.
Yin, J., Liu, D., Bao, L., Wang, Q., Chen, Y., Hou, S., Yue, Y, Yao, W and Gao, X. (2019). Tumor targeting and microenvironment-responsive multifunctional fusion protein for pro-apoptotic peptide delivery, Cancer letters, 452, 38-50. doi: 10.1016/j.canlet.2019.03.016.
Youmans, M. T. (2019). Identifying and Generating Candidate Antibacterial Peptides with Long Short-Term Memory Recurrent Neural Networks (Doctoral dissertation, Georgia Institute of Technology).