

Joshua Turner, Lizabeth Thomas, Sarah A. Kennedy
Department of Chemistry, Box 6949, Radford University, 801 E. Main St., Radford, VA 24141
ABSTRACT
Inhibiting the digestive enzyme α-glucosidase will slow the release of free glucose in the blood stream. This inhibition is one important strategy to treat sugar-related diseases such as diabetes. Since a 3-D structure of Saccharomyces cerevisiae α-glucosidase has not yet been solved, study of inhibition must be done through in silico creation of a structural model. Therefore, a new homology modeled structure of Saccharomyces cerevisiae α-glucosidase was built based on the most recent crystal structure of S. cerevisiae isomaltase (PDB: 3A47). This new model was used to dock five known natural α-glucosidase inhibitors to explore the putative allosteric drug binding pockets. Examination of the docking simulations and in silico mutagenesis revealed a potential druggable pocket for binding and a critical lysine residue capable of thermodynamically favorable binding with the inhibitors. In order to support the data analyzed from the docking simulation a series of inhibition assays were conducted on the wild-type enzyme and did show allosteric inhibition of α-glucosidase. These experiments contribute to a deeper understanding of the molecular level interactions required to inhibit this sugar metabolizing enzyme.
INTRODUCTION
α-glucosidase (EC 3.2.1.20) is a digestive enzyme which aids in the hydrolysis of α(1-4)-linked α-D-glucose molecules at the terminal end of polysaccharides (Jeske et. al., 2018). Inhibiting this protein slows the release of free glucose in the small intestines. The therapeutic advantage of inhibiting α-glucosidase is that it can be used to treat certain diseases associated with abnormally high blood glucose concentrations, such as Type II diabetes (diabetes mellitus). Those with diabetes mellitus do not secrete enough insulin following a meal, resulting in post-prandial hyperglycemia (Alberti and Zimmet, 1998). Inhibiting α-glucosidase can stop the body from entering a state of hyperglycemia by slowing the initial absorption of glucose in the blood stream, giving insulin a more manageable increase in blood-glucose levels (Robinson et al., 1991). There have been countless studies on inhibition of the active site in α-glucosidase using sugar mimics, which lead to the creation of Acarbose and Miglitol medications (Clissold and Edwards, 1988; Laube, 2002; Scott and Spencer, 2000; Sels et. al., 1999). However, these drugs induce side effects, including gastrointestinal pain, and constipation/diarrhea (Aoki et. al., 2010). The use of sugar mimicking drugs brings about the complication of interaction with other naturally occurring sugar-binding enzymes, which can lead to gastrointestinal distress.

Figure 1. Structures of flavonoids: (a) general flavonoid structure (b) quercetin with arrows pointing to the 3-OH of the C-ring (c) myricetin with arrows pointing to the 3-OH of the C-ring (d) luteolin.
The identification of natural allosteric inhibitors, which do not mimic sugars and bind to an area on the enzyme distinct from the active site, may reveal more potent inhibitors with fewer adverse effects; the inhibition studies of α-glucosidase in vitro and in vivo have created a pool of known and potential inhibitors with a specific flavonoid scaffold (Kim et. al., 2000; Singh et. al., 2014; Tadera et. al., 2006; Xu, 2010). Flavonoids consist of two specific chemical moieties: a benzopyran containing the A and C-rings, and a phenyl group referred to as the B-ring (Figure 1) (Panche et. al., 2016). Several flavonoids have previously been studied for inhibitory effects on α-glucosidase (Proença et al., 2017). Our goal is to identify and confirm how and where these compounds are binding to α-glucosidase as well as to verify their inhibitory mechanism because this will broaden the understanding of how allosteric inhibition of α-glucosidase works at the molecular level and inform future drug design. After investigating published research and compiling a list of natural inhibitors with IC50 values similar to acarbose, we decided on a series of flavonoids was identified for docking studies (Kim et al., 2000). These common inhibitors belong to a class of flavonoid derivatives isolated from various plant extracts. A derivative named luteolin even showed a higher potency than the current most effective drug acarbose (Kim et al., 2000). Flavonoids are classified into six groups based on the C-ring substituents and the linkage between the phenyl group and the benzopyran (Tadera et al., 2006). The flavonoids studied in this project fell into three categories: (1) flavonols (quercetin, myricetin, and fisetin), (2) flavanones (hesperetin, naringenin) and (3) flavones (luteolin) (Tadera et al., 2006). Here, we study these flavonoids for their ability to dock into allosteric positions on the enzyme, rather than to the active site where Acarbose and Miglitol bind.
Previously, researchers have studied the docking of luteolin, quercetin, and myricetin in a homology model created from the sequence of S. cerevisiae α-glucosidase and the structure of Bacillus cereus oligo-1,6-glucosidase (PBD: 1UOK) which, had a 38% amino acid sequence identity in common with yeast α-glucosidase (Bharatham et. al., 2008; Ferreira et al., 2010; Guerreiro et al., 2013). This docking study identified a hydrophobic, 14-residue pocket (pocket is defined as region on the biomolecule that is available for binding a molecule) and revealed that the inhibitory activity of luteolin was almost identical to the activities of quercetin and myricetin (Kim et al., 2000). In turn, it is thought that a 3-OH on the C-ring (Figure 1) of quercetin and myricetin is vital for holding the molecules in the most favorable orientation for inhibiting α-glucosidase. These results suggest that overall, the B-ring of flavones plays a more critical binding role than the A and C-rings in the inhibition of α-glucosidase. Homology models based on Bacillus cereus oligo-1,6-glucosidase (1UOK structure deposited in the protein data bank in 1998) were used in the majority of docking simulations for α-glucosidase until a recent docking study used a homolog based on the crystal structure of isomaltase from S. cerevisiae (PDB ID: 3AJ7) (Khan et al., 2014). This newer docking study, using 3AJ7, focused on biscoumarin analogs binding to the active site.
Our current study focuses on the allosteric binding of flavonoids to S. cerevisiae α-glucosidase based on the newest crystal structure of the isomaltase homologue (PDB 3A47). One of the impetuses for this work is the greatly improved sequence identity, which should afford creation of a more accurate model compared to other studies. Using a yeast isoform of α-glucosidase allows for easier in vivo testing and has proven to be a viable comparison to the human variants (Braun et. al., 1995). It is common practice in biochemistry to investigate many homologous enzymes from a variety of species to get a complete understanding of the relationship between structure and function.
To begin, we built and validated a homology model of S. cerevisiae α-glucosidase based on the newest crystal structures available. Next, this model was explored for druggable allosteric binding pockets. In silico docking studies provided molecular level information about drug-enzyme binding. Finally, the wild type S. cerevisiae α-glucosidase was inhibited in vivo, setting up the future investigation of these putative drug molecules.
METHODS
Creating an α-glucosidase protein model
S. cerevisiae is a common species used to study in vitro α-glucosidase inhibition and it is readily available for purchase. However, there is not a solved protein structure of S. cerevisiae α-glucosidase MAL12 (UniProtKB: P53341). To obtain a 3D structure of α-glucosidase to use as a scaffold to build our model, the primary sequence was entered in NCBI BLAST and results were restricted to sequences within the Protein Data Bank. The X-ray crystal structure of isomaltase from S. cerevisiae (PDB: 3A47) showed the highest similarity with 72% shared amino acid sequence identity. Using the sequence-template alignment and the full model builder features in ICM-Pro (Molsoft, LLC.), a homology model of the S. cerevisiae α-glucosidase MAL12 was built using the 3A47 structure as a template. The final model consisted of 586 amino acid residues and corresponds to amino acids 4-584 with two gaps in the α-glucosidase sequence at amino acids 282 and 563; in both gaps 3A47 had two additional residues.
Pockets, docking, and scoring
Using the feature ICM PocketFinder within the 3D predict section in ICM-Pro, seven pockets with likelihood of drug binding were located based on the factors of volume, area, hydrophobicity, and buriedness, which are some of the most common factors in considering binding capability. During structural analysis, maps were created to isolate the amino acids surrounding each of the seven potential druggable pockets. These in silico maps are defined regions of the molecule that are targeted during the docking studies. All potential inhibitors being docked were drawn into ICM-Pro using the 2D ligand edit feature and converted to a single chemical table for efficient docking to each pocket. The program then proceeds with docking the ligands into the pockets by placing the small molecule ligand within the defined 3D area of the enzyme pocket that was previously identified. The docking process allows in silico translational and rotational movement of both the amino acids in the enzyme pocket and the smaller ligand molecule. After sampling many structural fits, the program computes the best fit score based on allowable bond angles and intermolecular forces present. To obtain the best score, multiple trials were run to ensure the ligands docked in the most energetically favorable conformation. An average of four trials with a thoroughness setting of 5.0 was utilized for docking. The thoroughness setting ranges from 1 - 10 and affects how long it will dock each compound. Consultants at Molsoft advocated using settings of 1 - 5 with multiple runs to maximize time efficiency while still producing accurate results.
ICM-Pro creates a score for the likelihood that the small molecule will bind to the protein in an energetically favorable manner. This likelihood score is commonly used to compare the binding of a variety of small molecules or to compare wild-type and mutated pockets for binding ability. This score is analogous to a ΔG of binding with the following contributing factors: the number of rotatable torsions, hydrogen bond energy, hydrophobic energy, explicit van der Waals energy, internal conformation energy of the ligand, dissolved hydrogen bond donors and acceptors, electrostatic energy, and the mean force score. A strong score can vary by pocket; however, a score of -32 or smaller can be used if there is no known ligand to compare for binding in a specific pocket. With no proven inhibitors to the pockets in our new homology model, the baseline score of -32 was used in our study to signify an energetically favorable dock. To explore the composition of the most likely drug pockets, amino acids were mutated to test the importance of size, polarity, and acidity in drug binding. Default torsion parameters by ICM-Pro were used to position mutated amino acid in an energetically favorable manner.
Inhibitor assays
To support computational analysis of binding, a series of inhibition assays were conducted using S. cerevisiae α-glucosidase (MAL12 Sigma-Aldrich G5003) with the 4-nitrophenyl α-D-glucopyranoside substrate (Sigma-Aldrich N1377), which can absorb light at 405 nm when cleaved. Each assay was run in 100 µL reactions at 23 °C. Reaction components were added at the following concentrations: 5.214x10-3µM (10-2 U) enzyme, 50 mM sodium phosphate buffer (pH 7.0), and a substrate range of 25-500 µM. To allow for even drug dispersion in the solution, the inhibitors (luteolin, myricetin, quercetin) were suspended in DMSO, with a maximum of 5% DMSO per reaction well and tested at concentrations of 1 - 25 µM. The colorimetric reactions were monitored at 405 nm using a BIO-Tek PowerWave XS. The absorbances were recorded at intervals of 14 seconds, with a total read time of 20 minutes. Following data collection, kinetic curves were plotted and analyzed using Excel and Origin software.

Figure 2. NCBI BLAST alignment of the primary sequence of Saccharomyces cerevisiae isomaltase (PDB 3A47) with Saccharomyces cerevisiae α-glucosidase (Mal12).
RESULTS

Figure 3. Structural comparison of the S. cerevisiae α-glucosidase model (silver) with the isomaltase (PDB: 3A47) template (red). Areas of misalignment are highlighted with blue and green boxes.

Figure 4. Seven pockets identified in S. cerevisiae α-glucosidase that are potentially able to bind to small drug molecules. The darker shade of blue indicates the most common size drug pockets. The highest scoring pocket was Pocket 1 by both DLID and the size of the pocket. The second most drug-like pocket according to the DLID score is Pocket 3, which is an allosteric pocket. Pocket 2 also had a favorable score and was investigated in some initial docking studies.
The NCBI BLAST program scans all known protein sequences to find similar sequences to the query protein. In this case, the BLAST results showed that the two sequences S. cerevisiae α-glucosidase MAL12 and S. cerevisiae isomaltase (sequence from PDB structure 3A47, IMA1) aligned with 72% of their amino acids matching exactly and only five gaps out of a total of 587 residues (Figure 2). Uniprot has identified the nucleophile for the active site listed at amino acid D214 and the proton donor for the active site at E276, which were preserved in the alignment with 3A47. The tertiary structural alignment indicates a high degree of structural similarity with the template (Figure 3). There is some degradation in one α-helix (Figure 3, green outline) but this is distant from both the active site and the highest scoring allosteric pocket. As expected, there are also slight variations in some outer loop regions, which are typically more mobile (Figure 3, blue outline).
ICM PocketFinder located seven possible pockets with assigned scores that makes them likely for ligand binding, and their scores are within the range to be potential drug acceptors. The volume and area for drug-like binding pockets is marked by the blue cells in Figure 4 with the darker cells signifying a stronger possibility. The DLID column in Figure 4 indicates a drug-likelihood score. The highest scoring pocket was Pocket one by both DLID and the size of the pocket. Pocket one was confirmed to be the enzyme active site by comparing the pocket location to a model of isomaltase with an α-D-glucose bound into the structure (PDB: 3AXH). The second most drug-like pocket according to the DLID score is Pocket three, which is an allosteric pocket indicated by the yellow square on Figure 4. Pocket two also had a favorable score and was investigated in some initial docking studies (Figure 4, orange marker).
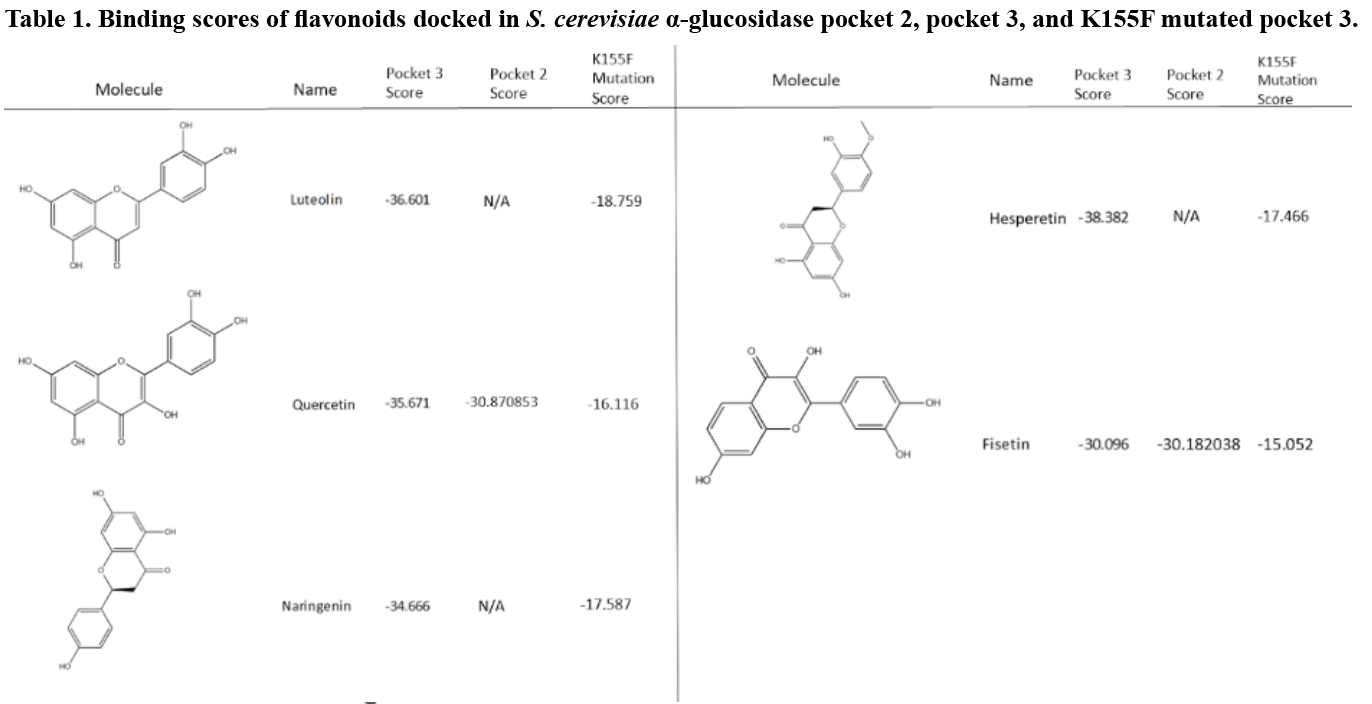

Figure 5. Homology model of S. cerevisiae α-glucosidase 3A47 (colored pink) binding pocket 2 electrostatic potential mesh viewed from the opening with docking of myricetin, the highest scoring flavonoid for this pocket.

Figure 6. Homology model of S. cerevisiae α-glucosidase (colored pink) binding pocket 3 electrostatic potential mesh view from the pocket opening with docking of hesperetin, the highest scoring flavonoid for this pocket.
Eight flavonoid derivatives, purchasable compounds known to inhibit ɑ-glucosidase, were docked into Pockets two and three. The compounds were then filtered by a score of -30 or lower, with the most negative being the most favorable. Pocket two bound to three of these compounds with scores close to -30: myricetin, quercetin, and fisetin (Table 1). The binding mode for each of these molecules varied greatly within the pocket. Hydrogen bonding was seen between multiple hydroxyl groups on the putative inhibitor and various amino acids side chain and backbone moieties. This is likely due to the location of the pocket in an area dominated by a loop structure (Figure 5). Thus, the binding mode for these inhibitors in Pocket two is not definitive.

Figure 7. Top scoring flavonoid derivatives docked into Pocket 3 (non-interactive amino acids removed for clarity). The hydrogen bond formed between the protein and inhibitor are shown.
When docking into Pocket three (Figure 6), five flavonoids (luteolin, quercetin, naringenin, hesperetin, and fisetin) scored below -30 (Table 1). Four of the compounds scored significantly below the -30 target with scores below -35, indicating a high likelihood of drug-like binding. Unlike Pocket two, there were key amino acids in Pocket three that appeared to play a significant role in the binding of the flavonoid. The flavonoid carbonyl group formed a hydrogen bond with the amino group at the end of K155 repeatedly during the docking (Figure 7). When this was coupled with 5C and 7C hydroxyl groups on the flavonoid to the backbone nitrogen of K236 and G160 respectively, binding was optimal.
To validate the importance of the K155 in the putative binding pocket, a K to F mutagenesis was performed on the model. The phenylalanine prevented the binding because the compounds could not interact with the key amino acids, thus altering the score above -20, which falls outside of the likely-druggable range (Table 1).

Figure 8. Inhibition of S. cerevisiae α-glucosidase by luteolin shown in a Lineweaver-Burk plot. Inhibition occurs even with sub-micromolar concentrations of inhibitor and appears to have a mixed or non-competitive binding mode.
An activity assay was conducted on α-glucosidase without inhibitor to verify our enzyme and substrate were functioning properly. The data retrieved from the assay was compiled and the first 120 seconds were used to calculate initial velocities. A Michaelis-Menten analysis of the data indicated an active enzyme with a Vmax of 0.00175 µM/s (S.E. = 5.29 x 10-5), a Km of 450 µM (S.E. = 23.28), and a Kcat of 0.3356 s-1. Next, DMSO concentrations were tested to ensure that adding this compound would not inhibit enzyme activity. An optimal 5% DMSO solution was required to solubilize the inhibitor molecules and did not interfere with native enzyme activity. Inhibition assays were conducted with luteolin (Figure 8) and myricetin to demonstrate their inhibitory properties, which indicated a trend of mixed/non-competitive inhibition of α-glucosidase.
DISCUSSION
The S. cerevisiae isomaltase was chosen for this modeling study because of its 72% identity match to S. cerevisiae α-glucosidase. Previous studies have used B. cereus oligo-1,6-glucosidase, which has only a 38% match (Bharatham et al., 2008). Despite α-glucosidase being a 1 - 4 linkage cleaving maltase, using a 1 - 6 cleaving isomaltase was more beneficial in preserving the structural accuracy due to the high sequence identity. Any active site variance is negligible considering all computational simulations were run on allosteric regions of the enzyme. Choosing S. cerevisiae isomaltase as the structural scaffold for our α-glucosidase model allowed us to improve sequence identity, model within the same species, and use the most current structural model.
The homology model created from isomaltase was used to study the docking of natural inhibitors known as flavonoids. Seven binding pockets were identified, one was the active site, and two were identified as potential druggable pockets. The binding of several flavonoids was shown to be the most thermodynamically favorable when docked into Pocket three, where a specific lysine residue was crucial for interacting with each inhibitor. This Pocket three lysine (K155) was mutated to a phenylalanine to see if this large, hydrophobic amino acid would block the compound’s access to that section of the pocket. This K to F mutation effectively blocked the primary carbonyl binding site and prevented interaction with the two amino acids important to binding with the inhibitor hydroxyl groups. These results indicate the importance of K155 in this drug-binding pocket.
Preliminary attempts at expressing S. cerevisiae α-glucosidase with the K155F mutation were not successful. However, the wild-type assays conducted with luteolin and myricetin displayed a similar mixed/non-competitive inhibition as reported in literature (Tadera et al., 2006). While this indicates the compounds allosterically inhibit α-glucosidase, the exact pocket location and the importance of the K155F residue must still be determined in vitro. To validate the importance of residue K155 from Pocket 3, next steps would include mutating this lysine in the S. cerevisiae α-glucosidase and performing inhibitory studies with wild-type and mutant proteins. Once validated, interactions with the specific lysine can be used to filter large compound libraries.
Docking simulations using large scale compound libraries play a key role in modern day drug discovery (Mario Geysen et. al., 2003). This study identifies an area on the S. cerevisiae α-glucosidase that could be used to inhibit the enzyme. Key amino acids within this druggable pocket have been identified and can be tested in vitro. Given this enzyme’s role in sugar metabolism, this study provides molecular-level information that can be used to develop better inhibitors that could potentially decrease side effects of current drugs Acarbose and Miglitol in treating diabetes mellitus. These inhibitors would slow the release of glucose monomers in the intestine, which would allow those diabetic patients better control over blood glucose levels.
REFERENCES
Alberti, K. G. and Zimmet, P. Z. (1998). Definition, diagnosis and classification of diabetes mellitus and its complications. Part 1: diagnosis and classification of diabetes mellitus provisional report of a WHO consultation. Diabetic Medicine : A Journal of the British Diabetic Association, 15(7), 539–553. https://doi.org/10.1002/(SICI)1096-9136(199807)15:7<539::AID-DIA668>3.0.CO;2-S.
Aoki, K., Muraoka, T., Ito, Y., Togashi, Y., and Terauchi, Y. (2010). Comparison of adverse gastrointestinal effects of acarbose and miglitol in healthy men: a crossover study. Internal Medicine (Tokyo, Japan), 49(12), 1085–1087. https://doi.org/10.2169/internalmedicine.49.3218.
Bharatham, K., Bharatham, N., Park, K. H., and Lee, K. W. (2008). Binding mode analyses and pharmacophore model development for sulfonamide chalcone derivatives, a new class of α-glucosidase inhibitors. Journal of Molecular Graphics and Modelling, 26(8), 1202–1212. https://doi.org/https://doi.org/10.1016/j.jmgm.2007.11.002.
Braun, C., Brayer, G. D., and Withers, S. G. (1995). Mechanism-based Inhibition of Yeast α-Glucosidase and Human Pancreatic α-Amylase by a New Class of Inhibitors. Journal of Biological Chemistry, 270(45), 26778–26781. https://doi.org/10.1074/JBC.270.45.26778.
Clissold, S. P., and Edwards, C. (1988). Acarbose. Drugs, 35(3), 214–243. https://doi.org/10.2165/00003495-198835030-00003.
Ferreira, S. B., Sodero, A. C. R., Cardoso, M. F. C., Lima, E. S., Kaiser, C. R., Silva, F. P., and Ferreira, V. F. (2010). Synthesis, biological activity, and molecular modeling studies of 1H-1,2,3-triazole derivatives of carbohydrates as α-glucosidases inhibitors. Journal of Medicinal Chemistry, 53(6), 2364–2375. https://doi.org/10.1021/jm901265h.
Guerreiro, L. R., Carreiro, E. P., Fernandes, L., Cardote, T. A. F., Moreira, R., Caldeira, A. T., Guedes, R. C., and Burke, A. J. (2013). Five-membered iminocyclitol α-glucosidase inhibitors: synthetic, biological screening and in silico studies. Bioorganic and Medicinal Chemistry, 21(7), 1911–1917. https://doi.org/10.1016/j.bmc.2013.01.030.
Jeske, L., Placzek, S., Schomburg, I., Chang, A., and Schomburg, D. (2018). BRENDA in 2019: a European ELIXIR core data resource. Nucleic Acids Research, 47(D1), D542–D549. https://doi.org/10.1093/nar/gky1048.
Khan, K. M., Rahim, F., Wadood, A., Kosar, N., Taha, M., Lalani, S., Khan, A., Fakhri, M. I., Junaid, M., Rehman, W., Khan, M., Perveen, S., Sajid, M., and Choudhary, M. I. (2014). Synthesis and molecular docking studies of potent α-glucosidase inhibitors based on biscoumarin skeleton. European Journal of Medicinal Chemistry, 81, 245–252. https://doi.org/10.1016/j.ejmech.2014.05.010.
Kim, J.-S., Kwon, C.-S., and Son, K. H. (2000). Inhibition of alpha-glucosidase and amylase by luteolin, a flavonoid. Bioscience, Biotechnology, and Biochemistry, 64(11), 2458–2461. https://doi.org/10.1271/bbb.64.2458.
Laube, H. (2002). Acarbose. Clinical Drug Investigation, 22(3), 141–156 https://doi.org/10.2165/00044011-200222030-00001.
Mario Geysen, H., Schoenen, F., Wagner, D., and Wagner, R. (2003). Combinatorial compound libraries for drug discovery: an ongoing challenge. Nature Reviews Drug Discovery, 2(3), 222–230. https://doi.org/10.1038/nrd1035.
Panche, A. N., Diwan, A. D., and Chandra, S. R. (2016). Flavonoids: an overview. Journal of Nutritional Science, 5, e47–e47. https://doi.org/10.1017/jns.2016.41.
Proença, C., Freitas, M., Ribeiro, D., Oliveira, E. F. T., Sousa, J. L. C., Tomé, S. M., Ramos, M. J., Silva, A. M. S., Fernandes, P. A., and Fernandes, E. (2017). α-Glucosidase inhibition by flavonoids: an in vitro and in silico structure-activity relationship study. Journal of Enzyme Inhibition and Medicinal Chemistry, 32(1), 1216–1228. https://doi.org/10.1080/14756366.2017.1368503.
Robinson, K. M., Begovic, M. E., Rhinehart, B. L., Heineke, E. W., Ducep, J.-B., Kastner, P. R., Marshall, F. N., and Danzin, C. (1991). New Potent α-Glucohydrolase Inhibitor MDL 73945 With Long Duration of Action in Rats. Diabetes, 40(7), 825 LP – 830. https://doi.org/10.2337/diab.40.7.825.
Scott, L. J., and Spencer, C. M. (2000). Miglitol: a review of its therapeutic potential in type 2 diabetes mellitus. Drugs, 59(3), 521–549. https://doi.org/10.2165/00003495-200059030-00012.
Sels, J.-P. J. E., Huijberts, M. S. P., and Wolffenbuttel, B. H. R. (1999). Miglitol, a new α-glucosidase inhibitor. Expert Opinion on Pharmacotherapy, 1(1), 149–156. https://doi.org/10.1517/14656566.1.1.149.
Singh, M., Kaur, M., and Silakari, O. (2014). Flavones: An important scaffold for medicinal chemistry. European Journal of Medicinal Chemistry, 84, 206–239. https://doi.org/https://doi.org/10.1016/j.ejmech.2014.07.013.
Tadera, K., Minami, Y., Takamatsu, K., and Matsuoka, T. (2006). Inhibition of alpha-glucosidase and alpha-amylase by flavonoids. Journal of Nutritional Science and Vitaminology, 52(2), 149–153. https://doi.org/10.3177/jnsv.52.149.
Xu, H. (2010). Inhibition kinetics of flavonoids on yeast α-glucosidase merged with docking simulations. Protein and Peptide Letters, 17(10), 1270–1279. https://doi.org/10.2174/092986610792231492.