

Miriam A. Daneff and Nafisa M. Jadavji
Department of Neuroscience, Carleton University, 1125 Colonel By Drive Ottawa, Ontario, Canada K1S 5B6
ABSTRACT
Cocaine is a widely abused illicit substance and has been gaining global popularity in recent years. It is understood that cocaine alters the normal functioning of the brain’s dopaminergic system to increase the amount of dopamine (DA) available to bind to receptors, subsequently potentiating DA’s euphoric effects. Knowledge regarding cocaine’s long-term addictive qualities is limited. Findings presented in this review demonstrate that DA D1 receptors (D1R) modulate the activity of the transcription factor ΔFosB, a marker of synaptic plasticity. Following cocaine use, dopaminergic activity is increased, subsequently causing the upregulation of ΔFosB. This upregulation of ΔFosB is correlated with an increase in expression of the N-methyl-d-aspartate receptor (NMDAR), which acts as a core component of long-term potentiation (LTP), a process underlying learning and memory. Thus, alterations to the normal synaptic functioning involved in learning and memory could explain the long-term impact of cocaine addiction, such as relapse. Based on the findings discussed in this review, further investigation of the role of these pathways in cocaine addiction is warranted.
INTRODUCTION
Addiction is a disease characterized by the repeated engagement of a behavior to achieve a temporary reward despite the negative consequences (Sussman and Sussman, 2011). Core components of addiction include tolerance and withdrawal (Gupta and Kulhara, 2007). Tolerance involves the need to engage in the behavior at a greater level to achieve the appetitive effects. Withdrawal refers to a dependence on the addictive behavior with physiological discomfort resulting from discontinuation of the behavior (Sussman and Sussman, 2011).
Addiction affects the functioning of brain reward-system circuitry (Gardner, 2011). The brain reward circuit, also referred to as the mesolimbic pathway, consists primarily of neuronal projections from the midbrain, specifically from the Ventral Tegmental Area (VTA) to the Nucleus Accumbens (NAc), followed by the transfer of this information to the prefrontal cortex (Figure 1) (Volkow and Morales, 2015). Midbrain dopaminergic connections projecting from the VTA are known for their role in reward processing and positive motivation (Bromberg-Martin et al., 2010). Interactions with these midbrain dopaminergic systems are thought to underlie the addictive properties of drugs, as preclinical studies generally show that drug abuse increases the functioning of the brain’s reward pathway (Gardner, 2011).
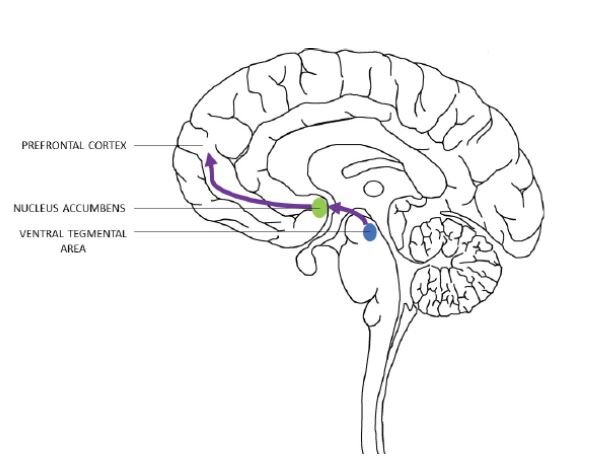
Figure 1. The flow of information in the brain reward pathway, from the ventral tegmental area to the nucleus accumbens (NA) to the prefrontal cortex (National Institute on Drug Abuse, 2016; Lynch, 2006).
Cocaine is a stimulant and an extremely addictive drug. Its effects at the synaptic level are extremely prominent in the NAc (Adinoff, 2004). Cocaine works by competitively blocking the dopamine transporter (DAT) located on the presynaptic neuron, thereby blocking the reuptake of DA and potentiating its reinforcing effects. This can cause users to feel euphoric and later continue chasing that high feeling (Beuming et al., 2008). The route of administration can also influence the addictive properties of cocaine as it determines the speed and duration of the high. For example, when smoking, the maximal concentration and effects of cocaine are attained almost instantly (Quenzer and Meyer, 2013). In contrast, intranasal administration produces a slower achievement of peak drug concentration as well as lower reports on the intensity of the high (Kiluk et al., 2013). Thus, these differences in the rate and magnitude of cocaine’s effects, as caused by the route of administration, contribute to the addictiveness of cocaine (Kiluk et al., 2013). Consistent with the general characteristics of drug addiction, cocaine users can experience tolerance as well as dependence on the drug, further contributing to cocaine’s addictive properties (Gardner, 2011).
The global cultivation of cocaine reached a peak in 2000, followed by a downward trend until 2013 (United Nations Office on Drugs and Crime, 2018). From 2015 to 2016, the global cocaine market underwent a significant expansion; global cultivation increased by 36%, global production by 25%, global drug seizures
by 23%, and the global number of users by 7% (United Nations Office on Drugs and Crime, 2018). In 2016, the worldwide prevalence of cocaine abuse was determined to be approximately 18.2 million people (United Nations Office on Drugs and Crime, 2018). The number of deaths involving cocaine abuse has also increased in recent years, further demonstrating the severity and growing level of cocaine abuse worldwide (United Nations Office on Drugs and Crime, 2018).

Figure 2. The release of DA from a presynaptic neuron, with arrival at DA D1 and D2 receptors on the postsynaptic neuron and DA reuptake by DAT on the presynaptic neuron (Quenzer and Meyer, 2013).
Currently, there is no accepted pharmacological treatment for cocaine addiction (Hernandez et al., 2018). Psychological therapies such as Cognitive Behavioral Therapy (CBT) are often the only method of treatment (Nestler, 2005). However, longterm characteristics of addiction, such as relapse, are pervasive and commonly observed in those seeking treatment for cocaine addiction. For example, a study from a substance abuse treatment center in the United States found that over 75% of individuals enrolled in a one-year treatment program for cocaine addiction relapse before being discharged (Sinha, 2011). Additional research is needed to better understand the neurobiological mechanisms underlying such long-term components of cocaine addiction. Research on synaptic plasticity and cocaine addiction provides novel insight to such factors.
Synaptic plasticity refers to the strengthening or weakening of synaptic transmission due to the activity or inactivity of neural circuits (Citri and Malenka, 2008). Synaptic plasticity is a core component of learning and memory (Takeuchi et al., 2014). Thus, drug-elicited changes in the normal functioning of synaptic plasticity may affect the processes involved in learning and memory (Lüscher and Malenka, 2011). The effects of drugs on synaptic plasticity could explain long-term characteristics of addiction, such as relapse (Kalivas and O’Brien, 2008). Therefore, future research is warranted to investigate the temporal implications of synaptic plasticity in cocaine addiction. Such research could provide insight for the future direction of potential pharmaceutical treatments. The aim of this paper is to provide an integrated and comprehensive overview on the role of synaptic plasticity in the context of cocaine addiction.
Mechanism of Cocaine in the Mesolimbic DA Pathway
Understanding cocaine’s mechanisms of action in the brain is paramount to research on potential pharmacological treatment for cocaine addiction. The dopamine transporter has been identified as the primary target of cocaine (X. Huang et al., 2009). It is a membrane-spanning protein located on the presynaptic neuron; the functioning of DAT is essential to the proper neurotransmission of DA (Block et al., 2015). The dopamine transporter is responsible for the removal of extracellular DA, transporting the neurotransmitter from the synapse back into the cytosol of the presynaptic neuron (Figure 2) (Vaughan and Foster, 2013). Preclinical studies utilizing DAT knockout mice demonstrate that cocaine is unable to hinder the clearance of synaptic DA in these rodents (Budygin et al., 2002; Giros et al., 1996; Jones et al., 1998). The inability of cocaine to affect the removal of DA in DAT knockout mice highlights the importance of DAT in cocaine’s mechanism of action in the brain.
Molecular studies seek to determine the location of the binding site of cocaine on DAT. The tertiary structure of DAT has not yet been characterized in detail; however, the structure of DAT’s bacterial homolog, the leucine transporter (LeuT), is well defined (Beuming et al., 2008). Thus, LeuT permits for homologous models of cocaine binding to be constructed (Beuming et al., 2008). Molecular research utilizing the LeuT homolog demonstrates that the initial binding sites of DA and cocaine on LeuT differ (X. Huang et al., 2009). Initially, cocaine binds to a site capable of accommodating it at a location that remains separate but near the DA binding site (X. Huang et al., 2009). Subsequently, conformational changes occur in the transporter that enable cocaine to move to the binding site of DA, thereby blocking DA from binding (X. Huang et al., 2009). Thus, this molecular evidence produced with the homologous LeuT model provides support for the competitive inhibition at the DAT binding site by cocaine.
Cocaine’s inhibition of DAT occurs primarily on presynaptic dopaminergic mesolimbic neurons (Kuhar, Ritz and Boja, 1991). Activation of the mesolimbic pathway is heavily implicated in the mediation of reward, with the activity of DA causing feelings of euphoria (Adinoff, 2004). Potentiation of DA’s euphoric effects by cocaine is thought to be the cause of the reinforcing and addictive properties of cocaine (Kuhar et al., 1991). Furthermore, the amount of extracellular DA is increased due to competitive inhibition of DAT (N. D. Volkow et al., 1997). The intensity of the high experienced by cocaine users is positively correlated with the rate at which cocaine blocks DAT (N D Volkow et al., 1999). Thus, cocaine’s mechanism of action in the mesolimbic pathway is at DAT; inhibition of DAT is responsible for prolonging DA’s effects, producing the feelings of euphoria typically experienced by users following cocaine administration.
ΔFosB in the Limbic System and Cocaine Addiction
In total, five DA receptors have been identified and labelled as D1-5 receptors (Tarazi, 2001). Dopamine D1 receptors (D1R) are located in the mesolimbic pathway and have been implicated in the pathophysiology of cocaine addiction (Self, 2014). For example, preclinical studies utilizing D1R knockout mice demonstrate a failure by these rodents to exhibit cocaine self-administration behavior (Self, 2014).
A member of the Fos family of transcription factors, ΔFosB, has been heavily implicated in drug addiction (Lobo et al., 2013; Nestler et al., 2001). Transcription factors such as ΔFosB are present inside of neurons and are responsible for activating or repressing gene expression (Vaquerizas et al., 2009). The ΔFosB transcription factor is located in small quantities in medium spiny neurons (MSN) in the NAc (Nestler, 2005). These MSN express many D1R that have been shown to modulate ΔFosB activity in the striatum (Moratalla et al., 1996; Muller and Unterwald, 2005). Acute administration of DA agonists, such as cocaine, increases levels of ΔFosB (Young et al., 1991). Experiments utilizing the administration of a selective D1R antagonist versus a selective D2R antagonist examine which DA receptor is responsible for the modulation of ΔFosB (Young et al., 1991). Antagonism of D1R resulted in lower levels of ΔFosB whereas antagonism of D2R had no effect on ΔFosB levels (Young et al., 1991). These results provide support for the hypothesis that D1Rs are linked to the regulation of ΔFosB.
The self-administration of cocaine produces a positive feedback loop, wherein sensitivity to cocaine’s reinforcing effects is propagated. Cocaine has a transient effect on limbic ΔFosB concentrations, mediated by the activation of mesolimbic D1R (Kelz et al., 1999). In the occurrence of chronic self-administration of cocaine, ΔFosB accumulates in the MSN of the NAc, persisting in large concentrations for long periods of time (Perrotti et al., 2008). As ΔFosB is a transcription factor, it is responsible for the regulation of gene expression (Vaquerizas et al., 2009). Therefore, ΔFosB is capable of inducing long-term molecular changes in the brain (Vaquerizas et al., 2009). Thus, researchers hypothesize that the accumulation of ΔFosB due to overactive D1R may underlie the mechanisms of prolonged sensitivity to cocaine (McClung et al., 2004; Nestler et al., 2001). In this way, a positive feedback loop is created, leading to continued cocaine use.
Preclinical studies utilizing transgenic rodents seek to investigate the implications of the accumulation of ΔFosB in cocaine administration. Mice that are engineered to overexpress ΔFosB display enhanced behavioral phenotypes when given cocaine (Kelz et al., 1999). For example, in comparison to wildtype mice, transgenic mice demonstrated an approximate 50% greater increase in locomotor activity when given cocaine (Kelz et al., 1999). Mice overexpressing ΔFosB demonstrate enhanced sensitivity to the reinforcing effects of cocaine, self-administering higher doses of the drug than the control mice (Kelz et al., 1999). Thus, the transcription factor ΔFosB may modulate the strength of the reinforcing effects of cocaine that are experienced by a user, thereby contributing to cocaine addiction.
The plasticity of cocaine’s effects on ΔFosB levels has been investigated through the use of western blot analysis in cocainedependent mice (Ehrlich et al., 2002). Results showed that limbic ΔFosB was upregulated in periadolescent mice but not in adult mice (Ehrlich et al., 2002). As young adolescent brains are still developing, molecular responses to psychostimulant drugs such as cocaine may differ from adult brain responses (Ehrlich et al., 2002). Thus, adolescents may be more vulnerable to cocaine addiction. These results highlight and provide an example of the brain’s unique plasticity and the role this plays in the regulation of cocaine’s effects on ΔFosB levels.
By increasing DA binding to D1R in the mesolimbic pathway, the administration of cocaine upregulates the activity of the transcription factor ΔFosB and subsequently alters gene expression. Upregulation of ΔFosB is correlated with an increase in sensitivity to the reinforcing effects of cocaine, thereby facilitating cocaine addiction. However, ΔFosB has a life span of only two months. Thus, the accumulation of ΔFosB alone cannot explain why recovering cocaine abusers often times relapse after months or years of abstinence (Nestler, 2005). Instead, long-term structural and functional changes that are induced by transcription factors such as ΔFosB may underlie the long-lasting effects of cocaine; this is known as synaptic plasticity (Kaste, 2009; Nestler, 2005).
Synaptic Plasticity and Cocaine Addiction
Synaptic plasticity can be defined as changes to the normal functioning of synaptic neurotransmission (Kauer and Malenka, 2007). Exposure to drugs such as cocaine can induce these longterm synaptic adaptations through modulation of the activity of molecular targets (Lüscher and Malenka, 2011). Researchers postulate that drug-evoked synaptic plasticity in the mesolimbic pathway results in long-term abnormalities in the functioning of reward circuitry (Kauer and Malenka, 2007; Yuan et al., 2013). The Neural Rejuvenation Hypothesis characterizes drug addiction in terms of synaptic plasticity mechanisms that are involved in learning and memory processes (Dong and Nestler, 2014). This hypothesis of drug addiction posits that addictive drugs act on dormant mechanisms that are normally responsible for the mediation of development, such as the mechanisms involved in learning and memory (Dong and Nestler, 2014). By hijacking these mechanisms, drugs are able to produce strong addiction-related memories, such as environmental cues with cocaine administration (Huang, et al., 2015). These memories are long-lasting and stable with mechanisms acting in the mesolimbic reward pathway (Huang et al., 2015). The activation of such addiction-related memories can lead to compulsive drug use, consequently increasing the likelihood of relapse (Goodman and Packard, 2016).
The formation of such strong, long-lasting memories is facilitated through a process known as long-term potentiation (LTP), a model of learning and memory that takes place at a synaptic level (Bliss and Collingridge, 1993). An essential component in the process of LTP is the glutamate receptor N-methyl-d-aspartate (NMDA) (Bliss and Collingridge, 1993). The presence of NMDA receptors (NMDAR) is vital in the initiation of LTP (Lüscher and Malenka, 2012; Zorumski and Izumi, 1998). Simultaneous activity between pre- and post-synaptic neurons triggers a large influx of calcium through NMDAR into the postsynaptic neuron (Lüscher and Malenka, 2012). Subsequently, multiple α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors (AMPAR) are inserted into the postsynaptic neuron’s membrane (Lüscher and Malenka, 2012). This increase in AMPAR expression is believed to underlie the strengthened synaptic transmission that is critical in LTP (Makino and Malinow, 2009).
As the Neural Rejuvenation Hypothesis postulates that cocaine “hijacks” learning and memory pathways, research seeks to investigate whether administration of cocaine results in abnormalities in LTP functioning. This has been examined through analysis of NMDAR activity (Mameli and Lüscher, 2011). Additionally, AMPAR activity has been investigated in the context of cocaine affecting LTP functioning, further providing support for the role of LTP in addiction (Mameli and Lüscher, 2011). Molecular studies seek to investigate NMDAR functioning at a transcriptional level. Results demonstrate that the transcription factor ΔFosB has a role in the expression of NMDAR in the mesolimbic pathway (Hiroi et al., 1998; McClung and Nestler, 2003). Upregulation of ΔFosB, such as that occurring due to longterm cocaine administration, has been implicated in the observed increase in NMDAR expression by MSN in the NAc (Dong and Nestler, 2014; Grueter et al., 2013). These results therefore demonstrate that cocaine increases NMDAR expression in the mesolimbic reward pathway through increased levels of synaptic DA, resulting in increased transcriptional activity of ΔFosB. As an increase in NMDAR expression is correlated with an increase in LTP, these results provide evidence suggesting that new addictionrelated memories may be formed following cocaine administration (Bliss and Collingridge, 1993). Therefore, this research indicates a possible mechanism underlying the long-lasting addictive behavior that is exhibited in cocaine abusers, such as that observed in certain recovered cocaine addicts who relapse after long periods of abstinence.
DISCUSSION
This review highlights the role of the dopaminergic system in the pathophysiology of cocaine addiction. Cocaine acts directly on the dopaminergic system, increasing levels of extracellular DA through competitive inhibition of DAT. This review discusses research on alterations in the functioning of the dopaminergic system that may be present in subjects with cocaine addiction.
Molecular studies have demonstrated that increases in the levels of synaptic DA due to cocaine have downstream effects underlying cocaine addiction. The transcription factor ΔFosB accumulates due to increases in DA binding to D1R following cocaine abuse. An increase in the concentration of ΔFosB is correlated with an increase in sensitivity to the reinforcing effects of cocaine. Thus, upregulation of ΔFosB due to increases in synaptic DA influences cocaine addiction. The preclinical study examined in this review by Kelz and colleagues (1999) produced results in accordance with these findings. This study, however, utilized adult mice only. As discussed previously, the regulation of ΔFosB in response to cocaine can differ between cocainedependent adult and periadolescent mice (Ehrlich et al., 2002). Thus, future research may benefit from investigating the effects of overexpression of ΔFosB in periadolescent mice.
Increased ΔFosB also correlates with an increase in NMDAR expression, a core component of LTP. Thus, by increasing the expression of NMDAR, cocaine alters the normal synaptic functioning involved in learning and memory processes. This cocaine-induced synaptic plasticity can consequently produce long-lasting, addiction-related memories. Thus, synaptic plasticity due to cocaine could explain symptoms of cocaine addiction such as cue-related cravings and relapse.
A comprehensive understanding of the neural mechanisms underlying cocaine addiction is vital in the research for novel treatments of cocaine addiction. The implications of synaptic plasticity in the pathophysiology of cocaine addiction, as discussed in this review, provide promise and direction for future
research in pharmacological treatment for this disease. Based on literature discussed in this review, research investigating the effectiveness of the inhibition of ΔFosB or NMDAR in cocaine addiction is justified. For example, antagonists of ΔFosB that inhibit its binding to target genes could aid in preventing cocaineinduced, altered gene expression (Wang et al., 2012). Antagonists of NMDAR could aid in inhibiting cocaine-induced LTP, thereby impeding the formation of addiction-related memories (Bisaga and Popik, 2000).
In addition to pharmaceutical treatments, the role of synaptic plasticity in the pathophysiology of cocaine addiction provides insight for the direction of psychological therapies. Contingency management (CM) is a psychotherapy treatment used for substance abuse (Rash et al., 2017). The use of CM treatment in conjunction with other psychological interventions has demonstrated the ability to increase cocaine abstinence (Schierenberg et al., 2012). Contingency management utilizes the principles of operant conditioning, a method of learning, to increase desired behavior (Trotman and Taxman, 2011). As demonstrated with the Neural Rejuvenation Hypothesis, cocaine, through its effects on LTP, alters the normal functioning of learning. Thus, psychotherapies such as CM that target learning processes may aid in the creation of new behaviors that replace cocaine-learned behaviors. Therefore, this review provides support for the research and use of psychotherapies to target learning processes.
This review provides evidence for the role of synaptic plasticity in the pathophysiology of cocaine addiction. Given that cocaine is a highly addictive drug of abuse with severe consequences and no currently-approved pharmacological treatment, continued research on the pathophysiology and treatment of cocaine addiction is vital.
REFERENCES
Adinoff, B. (2004). Neurobiologic processes in drug reward and addiction. Harvard Review of Psychiatry, 12(6), 305–320, available: https://doi.org/10.1080/10673220490910844.
Beuming, T., Kniazeff, J., Bergmann, M. L., Shi, L., Gracia, L., Raniszewska, K., Hauck Newman, A., Javitch, J. A., Weinstein, H., Gether, U., Loland, C. J. (2008). The binding sites for cocaine and dopamine in the dopamine transporter overlap. Nature Neuroscience, 11(7), 780–789, available: https://doi.org/10.1038/nn.2146.
Bisaga, A., and Popik, P. (2000). In search of a new pharmacological treatment fo rdrug and alcohol addiction: N-methyl-d-aspartate (NMDA) antagonists. Drug and Alcohol Dependence, 59(1), 1–15, available: https://doi.org/10.1016/S0376-8716(99)00107-6.
Bliss, T. V. P., and Collingridge, G. L. (1993). A synaptic model of memory: longterm potentiation in the hippocampus. Nature, 361(6407), 31–39, available: https://doi.org/10.1038/361031a0.
Block, E. R., Nuttle, J., Balcita-Pedicino, J. J., Caltagarone, J., Watkins, S. C., Sesack, S. R., Sorkin, A. (2015). Brain Region-Specific Trafficking of the Dopamine Transporter. Journal of Neuroscience, 35(37), available: https://doi.org/10.1523/JNEUROSCI.1391-15.2015.
Bromberg-Martin, E. S., Matsumoto, M., Hikosaka, O. (2010). Dopamine in motivational control: rewarding, aversive, and alerting. Neuron, 68(5), 815–834, available: https://doi.org/10.1016/j.neuron.2010.11.022.
Budygin, E. A., John, C. E., Mateo, Y., Jones, S. R. (2002). Lack of cocaine effect on dopamine clearance in the core and shell of the nucleus accumbens of dopamine transporter knock-out mice. The Journal of Neuroscience : The Official Journal of the Society for Neuroscience, 22(10), RC222, available:https://doi.org/10.1523/JNEUROSCI.22-10-j0002.2002.
Citri, A., and Malenka, R. C. (2008). Synaptic Plasticity: Multiple Forms, Functions and Mechanisms. Neuropsychopharmacology, 33(1), 18–41, available: https://doi.org/10.1038/sj.npp.1301559.
Dong, Y., and Nestler, E. J. (2014). The neural rejuvenation hypothesis of cocaine addiction. Trends in Pharmacological Sciences, 35(8), 374–383, available: https://doi.org/10.1016/j.tips.2014.05.005.
Ehrlich, M. E., Sommer, J., Canas, E., Unterwald, E. M. (2002). Periadolescent mice show enhanced DeltaFosB upregulation in response to cocaine and amphetamine. The Journal of Neuroscience : The Official Journal of the Society for Neuroscience, 22(21), 9155–9159, available: https://doi. org/10.1523/JNEUROSCI.22-21-09155.2002.
Gardner, E. L. (2011). Addiction and brain reward and antireward pathways. Advances in Psychosomatic Medicine, 30, 22–60, available: https://doi.org/10.1159/000324065.
Giros, B., Jaber, M., Jones, S. R., Wightman, R. M., Caron, M. G. (1996). Hyperlocomotion and indifference to cocaine and amphetamine in mice lacking the dopamine transporter. Nature, 379(6566), 606–612, available: https://doi.org/10.1038/379606a0.
Goodman, J. and Packard, M. G. (2016). Memory systems and the addicted brain. Frontiers in Psychiatry, 7, 24, available: https://doi.org/10.3389/fpsyt.2016.00024.
Grueter, B. A., Robison, A. J., Neve, R. L., Nestler, E. J., Malenka, R. C. (2013). FosB differentially modulates nucleus accumbens direct and indirect pathway function. Proceedings of the National Academy of Sciences, 110(5), 1923–1928, available: https://doi.org/10.1073/pnas.1221742110.
Gupta, S. and Kulhara, P. (2007). Cellular and molecular mechanisms of drug dependence: An overview and update. Indian Journal of Psychiatry, 49(2), 85–90, available: https://doi.org/10.4103/0019-5545.33253.
Hernandez, N. S., Ige, K. Y., Mietlicki-Baase, E. G., Molina-Castro, G. C., Turner, C. A., Hayes, M. R., Schmidt, H. D. (2018). Glucagon-like peptide-1 receptor activation in the ventral tegmental area attenuates cocaine seeking in rats. Neuropsychopharmacology, 43(10), 2000–2008, available: https://doi.org/10.1038/s41386-018-0010-3.
Hiroi, N., Marek, G. J., Brown, J. R., Ye, H., Saudou, F., Vaidya, V. A., Duman, R. S., Greenberg, M. E., Nestler, E. J. (1998). Essential role of the fosB gene in molecular, cellular, and behavioral actions of chronic electroconvulsive seizures. The Journal of Neuroscience : The Official Journal of the Society for Neuroscience, 18(17), 6952–6962, available: https://doi.org/10.1523/JNEUROSCI.18-17-06952.1998.
Huang, X., Gu, H. H., Zhan, C.-G. (2009). Mechanism for cocaine blocking the transport of dopamine: insights from molecular modeling and dynamics simulations. The Journal of Physical Chemistry. B, 113(45), 15057–15066, available: https://doi.org/10.1021/jp900963n.
Huang, Y. H., Schlüter, O. M., Dong, Y. (2015). Silent Synapses Speak Up: Updates of the Neural Rejuvenation Hypothesis of Drug Addiction. The Neuroscientist: A Review Journal Bringing Neurobiology, Neurology and Psychiatry, 21(5), 451–459, available: https://doi.org/10.1177/1073858415579405.
Jones, S. R., Gainetdinov, R. R., Jaber, M., Giros, B., Wightman, R. M., Caron, M. G. (1998). Profound neuronal plasticity in response to inactivation of the dopamine transporter. Proceedings of the National Academy of Sciences of the United States of America, 95(7), 4029–4034, available: http://www.ncbi. nlm.nih.gov/pubmed/9520487.
Kalivas, P. W. and O’Brien, C. (2008). Drug Addiction as a Pathology of Staged Neuroplasticity. Neuropsychopharmacology, 33(1), 166–180, available: https://doi.org/10.1038/sj.npp.1301564.
Kaste, K. (2009). Transcription Factors ΔFosB and CREB in Drug Addiction: Studies in Models of Alcohol Preference and Chronic Nicotine Exposure University of Helsinki, available: https://pdfs.semanticscholar.org/4bbd/c6b798a1ad96480db2009eaca91b29a771ca.pdf.
Kauer, J. A. and Malenka, R. C. (2007). Synaptic plasticity and addiction. Nature Reviews Neuroscience, 8(11), 844–858, available: https://doi.org/10.1038/nrn2234.
Kelz, M. B., Chen, J., Carlezon, W. A., Whisler, K., Gilden, L., Beckmann, A. M., Steffen, C., Zhang, Y., Marotti, L., Self, D. W., Tkatch, T., Baranauskas, G., Surmeier, D. J., Neve, R. L., Duman, R. S., Picciotto, M. R., Nestler, E. J. (1999). Expression of the transcription factor ΔFosB in the brain controls sensitivity to cocaine. Nature, 401(6750), 272–276, available: https://doi.org/10.1038/45790.
Kiluk, B. D., Babuscio, T. A., Nich, C., Carroll, K. M. (2013). Smokers versus snorters: do treatment outcomes differ according to route of cocaine administration? Experimental and Clinical Psychopharmacology, 21(6), 490–498, available: https://doi.org/10.1037/a0034173.
Kuhar, M. J., Ritz, M. C., Boja, J. W. (1991). The dopamine hypothesis of the reinforcing properties of cocaine. Trends in Neurosciences, 14(7), 299–302, available: https://doi.org/10.1016/0166-2236(91)90141-G.
Lobo, M. K., Zaman, S., Damez-Werno, D. M., Koo, J. W., Bagot, R. C., DiNieri, J. A., Nugent, A., Finkel, E., Chaudhury, D., Chandra, R., Riberio, E., Rabkin, J., Mouzon, E., Cachope, R., Cheer, J. F., Han, M., Dietz, D. M., Self, D. W., Hurd, Y. L., Vialou, V., Nestler, E. J. (2013). ΔFosB induction in striatal medium spiny neuron subtypes in response to chronic pharmacological, emotional, and optogenetic stimuli. The Journal of Neuroscience : The Official Journal of the Society for Neuroscience, 33(47), 18381–18395, available: https://doi.org/10.1523/JNEUROSCI.1875-13.2013.
Lüscher, C. and Malenka, R. C. (2011). Drug-Evoked Synaptic Plasticity in Addiction: From Molecular Changes to Circuit Remodeling. Neuron, 69(4), 650–663, available: https://doi.org/10.1016/j.neuron.2011.01.017.
Lüscher, C. and Malenka, R. C. (2012). NMDA receptor-dependent long-term potentiation and long-term depression (LTP/LTD). Cold Spring Harbor Perspectives in Biology, 4(6), available: https://doi.org/10.1101/cshperspect.a005710.
Makino, H. and Malinow, R. (2009). AMPA receptor incorporation into synapses during LTP: the role of lateral movement and exocytosis. Neuron, 64(3), 381–390, available: https://doi.org/10.1016/j.neuron.2009.08.035.
Mameli, M. and Lüscher, C. (2011). Synaptic plasticity and addiction: Learning mechanisms gone awry. Neuropharmacology, 61(7), 1052–1059, available: https://doi.org/10.1016/j.neuropharm.2011.01.036.
McClung, C. A. and Nestler, E. J. (2003). Regulation of gene expression and cocaine reward by CREB and ΔFosB. Nature Neuroscience, 6(11), 1208–1215, available: https://doi.org/10.1038/nn1143.
McClung, C. A., Ulery, P. G., Perrotti, L. I., Zachariou, V., Berton, O., Nestler, E. J. (2004). ΔFosB: a molecular switch for long-term adaptation in the brain. Molecular Brain Research, 132(2), 146–154, available: https://doi.org/10.1016/j.molbrainres.2004.05.014.
Moratalla, R., Vallejo, M., Elibol, B., Graybiel, A. M. (1996). D1-class dopamine receptors influence cocaine-induced persistent expression of Fos-related proteins in striatum. Neuroreport, 8(1), 1–5, available: https://doi.org/10.1097/00001756-199612200-00001.
Muller, D. L. and Unterwald, E. M. (2005). D1 Dopamine Receptors Modulate FosB Induction in Rat Striatum after Intermittent Morphine Administration. Journal of Pharmacology and Experimental Therapeutics, 314(1), 148–154, available: https://doi.org/10.1124/jpet.105.083410.
Nestler, E J, Barrot, M., Self, D. W. (2001). DeltaFosB: a sustained molecular switch for addiction. Proceedings of the National Academy of Sciences of the United States of America, 98(20), 11042–11046, available: https://doi.org/10.1073/pnas.191352698.
Nestler, Eric J. (2005). The neurobiology of cocaine addiction. Science & Practice Perspectives / a Publication of the National Institute on Drug Abuse, National Institutes of Health, 3(1), 4–10, available: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2851032/pdf/spp-03-1-4.pdf.
Perrotti, L. I., Weaver, R. R., Robison, B., Renthal, W., Maze, I., Yazdani, S., Elmore, R. G., Knapp, D. J., Selley, D. E., Martin, B. R., Sim-Selley, L., Bachtell, R. K., Self, D. W., Nestler, E. J. (2008). Distinct patterns of ΔFosB induction in brain by drugs of abuse. Synapse, 62(5), 358–369, available: https://doi.org/10.1002/syn.20500.
Quenzer, L. and Meyer, J. (2013). Psychopharmacology: Drugs, the Brain, and Behaviour (Second). Sunderland: Sinauer Associates, available: https://www.sinauer.com/media/wysiwyg/tocs/Psychopharmacology2.pdf.
Rash, C. J., Stitzer, M., Weinstock, J. (2017). Contingency Management: New Directions and Remaining Challenges for An Evidence-Based Intervention. Journal of Substance Abuse Treatment, 72, 10–18, available: https://doi.org/10.1016/j.jsat.2016.09.008.
Schierenberg, A., van Amsterdam, J., van den Brink, W., Goudriaan, A. E. (2012). Efficacy of contingency management for cocaine dependence treatment: a review of the evidence. Current Drug Abuse Reviews, 5(4), 320–331, available: https://doi.org/10.2174/1874473711205040006.
Self, D. W. (2014). Diminished role for dopamine D1 receptors in cocaine addiction? Biological Psychiatry, 76(1), 2–3, available: https://doi.org/10.1016/j.biopsych.2014.04.006.
Sinha, R. (2011). New findings on biological factors predicting addiction relapse vulnerability. Current Psychiatry Reports, 13(5), 398–405, available: https://doi.org/10.1007/s11920-011-0224-0.
Sussman, S. and Sussman, A. N. (2011). Considering the definition of addiction. International Journal of Environmental Research and Public Health, 8(10), 4025–4038, available: https://doi.org/10.3390/ijerph8104025.
Takeuchi, T., Duszkiewicz, A. J., Morris, R. G. M. (2014). The synaptic plasticity and memory hypothesis: encoding, storage and persistence. Philosophical Transactions of the Royal Society of London. Series B, Biological Sciences, 369(1633), 20130288, available: https://doi.org/10.1098/rstb.2013.0288.
Tarazi, F. I. (2001). Neuropharmacology of dopamine receptors:: Implications in neuropsychiatric diseases. Journal for Scientific Research. Medical Sciences, 3(2), 93–104, available: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3174705/pdf/squmj-03-93.pdf.
Trotman, A. J. and Taxman, F. S. (2011). Implementation of a Contingency Management-Based Intervention in a Community Supervision Setting: Clinical Issues and Recommendations. Journal of Offender Rehabilitation, 50(5), 235–251, available: https://doi.org/10.1080/10509674.2011.585924.
UNODC. (2018). Analysis of Drug Markets: Opiates, cocaine, cannabis, synthetic drugs, available: https://www.unodc.org/wdr2018.
Vaquerizas, J. M., Kummerfeld, S. K., Teichmann, S. A., Luscombe, N. M. (2009). A census of human transcription factors: function, expression and evolution. Nature Reviews Genetics, 10(4), 252–263, available: https://doi.org/10.1038/nrg2538.
Vaughan, R. A. and Foster, J. D. (2013). Mechanisms of dopamine transporter regulation in normal and disease states. Trends in Pharmacological Sciences, 34(9), 489–496, available: https://doi.org/10.1016/j.tips.2013.07.005.
Volkow, N. D., Wang, G.-J., Fischman, M. W., Foltin, R. W., Fowler, J. S., Abumrad, N. N., S. Vitkun, Logan, J., Gatley, S. J., Pappas, N., Hitzemann, R., Shea, C. E. (1997). Relationship between subjective effects of cocaine and dopamine transporter occupancy. Nature, 386(6627), 827–830, available: https://doi.org/10.1038/386827a0.
Volkow, N D, Fowler, J. S., Wang, G. J. (1999). Imaging studies on the role of dopamine in cocaine reinforcement and addiction in humans. Journal of Psychopharmacology (Oxford, England), 13(4), 337–345, available: https://doi.org/10.1177/026988119901300406.
Volkow, N. D. and Morales, M. (2015). The Brain on Drugs: From Reward to Addiction. Cell, 162(4), 712–725, available: https://doi.org/10.1016/j.cell.2015.07.046.
Wang, Y., Cesena, T. I., Ohnishi, Y., Burger-Caplan, R., Lam, V., Kirchhoff, P. D., Larsen, S. D., Larsen, M. J., Nestler, E. J., Rudenko, G. (2012). Small molecule screening identifies regulators of the transcription factor ΔFosB. ACS Chemical Neuroscience, 3(7), 546–556, available: https://doi.org/10.1021/cn3000235.
Young, S. T., Porrino, L. J., Iadarola, M. J. (1991). Cocaine induces striatal c-fosimmunoreactive proteins via dopaminergic D1 receptors. Proceedings of the National Academy of Sciences of the United States of America, 88(4), 1291–1295, available: https://doi.org/10.1073/pnas.88.4.1291.
Yuan, T., Mameli, M., O’Connor, E. C., O’ Connor, E. C., Dey, P. N., Verpelli, C., Sala, C., Perez-Otano, I., Luscher, C., Bellone, C. (2013). Expression of cocaine-evoked synaptic plasticity by GluN3A-containing NMDA receptors. Neuron, 80(4), 1025–1038, available: https://doi.org/10.1016/j.neuron.2013.07.050.
Zorumski, C. F. and Izumi, Y. (1998). Chapter 12 Modulation of LTP induction by NMDA receptor activation and nitric oxide release. Progress in Brain Research, 118, 173–182, available: https://doi.org/10.1016/S0079-6123(08)63207-0.