
Author: Amanda R. Albanese and Rebeka R. Merson
Institution: Rhode Island College
Date: November 2008
Abstract
β-Actin (ACTB) is a ubiquitously expressed cytoskeletal protein. Involved in cell movement and structure, it is usually expressed at constant levels within the cell. We have successfully cloned and sequenced ACTB cDNA from little skate (Leucoraja erinacea) from the 5' untranslated region through the poly-adenylated tail. The deduced protein was 374 amino acids in length, with a 99.7% identity to the human ortholog of ACTB. ACTB is highly conserved among vertebrates and is used as a protein loading control in Western blots and other experiments in which heterogeneous samples are used. In recent years, ACTB has been used as a reference gene in quantitative real-time polymerase chain reaction (qPCR) studies where it is used as a control for amplification variations between samples. Little skate (Leucoraja erinacea) is an emerging bioindicator species for toxicology and a biomedical model for reproductive endocrinology; however, limited genomic information from little skates is available. Genome sequencing projects in nontraditional animal models is providing new data in the fields of toxicogenomics, bioinformatics and the development of molecular tools. Therefore, in this experiment, ACTB cDNA was amplified and cloned from little skate in order to construct a standard curve for future qPCR data analysis.
Introduction
The six actin isoforms are proteins involved in cell motility, structure, and integrity. Actins are found in all eukaryotic cells and are highly conserved among vertebrates. The β-actin isoform (ACTB) is one of the two nonmuscle cytoskeletal actins, which act as a building block of cellular microfilaments (Ng et al. 1985). ACTB contains three conserved domains: the ATP binding site, the gelsolin binding site, and the profilin binding site. ACTB exhibits ATPase activity in order to hydrolyze ATP for filament assembly. This conserved site is found in the promoter of ACTB where it binds one molecule of ATP and either calcium or magnesium ions. Gelsolin and profilin are barbed-end binding proteins. These proteins cap the end of actin polymers, stabilizing the end of the protein and helping to regulate polymerization (Marchler-Bauer et al. 2007).
We will use ACTB as a reference gene to normalize data for quantitative real-time PCR (qPCR) analysis. It is often used as a protein loading control in Western blots and as a control in other experiments in which heterogeneous samples are used. Other commonly used reference genes include 18S ribosomal RNA (18S rRNA), ribosomal protein 18 (rp18), elongation factor 1 alpha (ef1a), glucose-6-phosphate dehydrogenase (g6pd), glyceraldehydes-3-phosphate dehydrogenase (gapdh), hypoxanthine phosphoibosyltransferase 1 (hprt1), and tata box binding protein (tbp) (Bustin 2005).
Quantitative real-time PCR (qPCR) is a technique used to accurately and precisely amplify and quantify nucleic acid molecules. The fluorescent signal of the reporter dye (SYBR Green) increases proportionally to the quantity of double stranded DNA present in the reaction. By measuring the increase in fluorescence with increasing PCR cycle number, the specific amount of a DNA amplicon can be quantified. The starting quantity of the amplified target DNA molecule is inversely proportional to the threshold cycle (Ct). This refers to the cycle at which the fluorescent signal of the sample has increased above the background fluorescence level. The Ct is measured during the most efficient portion of a PCR, in which two copies of the amplicon are generated per cycle. This is known as the exponential phase. A standard curve generated with multiple standards of known starting quantities is used to extrapolate the starting quantities in unknown samples. The ACTB starting quantity values of these unknown samples will be used for normalization of gene expression data from the experimental gene to be studied. Selection and optimization of a reference gene is an extremely important step in the development of a successful, reproducible qPCR assay (Bustin 2005).
Little skates (Leucoraja erinacea) are benthic marine fishes from the class Chondrichthyes. Used for the study of reproductive biology, embryonic development and endocrinology, little skate is also an emerging bioindicator species for toxicology (Koob and Callard 1999). Aquatic animal models, such as little skate, are easily exposed to waterborne pollutants through the surface of the gills and skin as well as through the consumption of contaminated prey. This allows for easy study of the effects of these compounds on physiological processes. Fish biological models are advantageous for use in research because of their high fecundity, externally fertilized eggs, and relatively brief generation times (Schmale et al. 2006). Data from the Northeast Fisheries Science Council (NEFSC) indicates that over the past few years, population levels of little skate have increased (NOAA 2006).
Little skates possess molecular systems for dealing with salt and water homeostasis, cell volume regulation, and environmental and internal osmotic sensing. These well developed systems are similar to those found in humans. Thus, little skates have become important models in studies of transport-related diseases such as cystic fibrosis and xenobiotic transport. The limited genomic information available hinders further research in a number of biomedical fields. Genomic information from little skate will be useful in the field of toxicogenomics, the study of an animal's genome to characterize toxicologically relevant genes and to understand the genetic basis for various diseases (Ballatori et al. 2003). Work to completely sequence the little skate genome is currently in process through the Mount Desert Island Biological Institute (MDIBL) (Mattingly et al. 2004).
Materials and Methods
Tissue Sample Collection
Skates were sampled by biologists from the Woods Hole Oceanographic Institution. 1 - 2 g tissue samples dissected for PCR analysis were immediately stored in RNALater (Ambion, Foster City, CA) and stored at -20°C or -80°C until processed.
Total RNA Isolation and cDNA synthesis
Total RNA was extracted from 100 - 200 mg liver, intestine, stomach and pancreas samples using the Invitrogen Purelink Micro to Midi Total RNA extraction Kit (Carlsbad, CA) or the Qiagen RNAeasy Mini Kit (Valencia, CA). These tissue types were selected because they were the subject of additional experiments with different target genes. The manufacturer's protocol was followed with the addition of a 20 minute digestion with 65 μl of 20 mg/ml proteinase K after homogenization in the lysis solution in the intestine tissue samples. The total RNA was eluted from the column three times with 30 μl, 50 μl, and 50 μl of DNAse, RNAse free water (5 Prime, Gaithersburg, MD) and the concentration measured with a Nanodrop spectrophotometer. The quality of the RNA was assessed through the visualization of the 18S and 28S rRNA subunits on a RNA denaturing gel. RNA samples were stored at -80 °C for further use.
cDNA was synthesized from 3.5 μg total RNA using the Sigma-Aldrich eAMV reverse transcription kit (St. Louis, MO). The manufacturer's protocol was followed using an optimized incubation temperature for little skate total RNA of 55 °C. To obtain the 5' and 3' ends of the sequence, RACE Ready cDNA was generated with 1 mg of total RNA using the Clontech SMART RACE cDNA Amplification Kit (Mountain view, CA).
Primer Design
Gene specific primers (Table 1) targeting ACTB cDNA were designed based on the published sequences in GenBank from zebrafish (Danio rerio, AAH45879), human (Homo sapiens, BAD96645), and leopard shark (Triakis semifasciata, AB084472). A multiple sequence alignment was generated using MacVector software and primers were designed in conserved regions of the nucleotide sequence. Forward and reverse primers were designed with similar melting temperatures to optimize PCR annealing temperatures.

Table 1. ACTB Primer and PCR condition details. UPM, universal primer mix, NUP, nested universal primer (SMART RACE, Clontech)
Rapid Amplification of cDNA Ends (RACE) primers (Table 1) were designed to amplify the 5' and 3' ends of the ACTB sequence based on the sequencing results of the initial ACTB fragment. Primers were designed based on the isolated liver ACTB sequence. The sequence obtained from the RACE experiments with pancreas cDNA was identical where it overlapped the initial fragment cloned and sequenced from liver cDNA (5' RACE 46 bp overlap, 3' RACE 146 bp overlap). Alternative splicing has not been recorded for ACTB allowing for the use of multiple tissue types for initial isolation of ACTB and for the RACE reactions (NCBI GENE Entry Maglott et al 2005).
qPCR primers (Table 1) were designed inside the resulting sequence of the cDNA construct spanning predicted exon-exon junctions from human (BAD96645) and zebrafish (AAH45879) genomes producing a 138 base pair amplicon. Using the GCG Wisconsin Package maintained by the RI-INBRE Bioinformatics Core at the University of Rhode Island, a visual diagram of predicted secondary structures was generated. These areas of the molecule were avoided in the design of the target amplicon so that the primers were designed in an area where binding would not be hindered.
PCR
We amplified the initial ACTB cDNA sequence using Finnzyme Phusion polymerase with GC Buffer (New England Biolabs, Beverly, MA) with cDNA from liver and intestine (see Table 1 for PCR cycling parameters). To optimize the PCR conditions, four different annealing temperatures were tested as a gradient to isolate ACTB. All PCR products were resolved by on a 1.5% agarose gel containing ethidium bromide and visualized by UV illumination. Based on the position of the initial ACTB primers, after gel analysis, a fragment approximately 1150 base pairs was isolated from the liver cDNA sample at 66 °C for further cloning studies.
RACE reactions
5' and 3' RACE Ready pancreas cDNA was amplified to isolate the 5' untranslated region and the 3' end of little skate ACTB using the Clontech SMART RACE cDNA Amplification Kit. Primary and nested reactions were performed over a 5 temperature gradient to isolate the sequences (Table 1). Control reactions containing only one of the two primers were performed to check for nonspecific binding. RACE products were subjected to gel electrophoresis on a 1% agarose gel for visualization of the expected sequences. The RACE primers generated 5' untranslated region that was approximately 250 base pairs in length and the 3' RACE product was approximately 700 base pairs in length.
cDNA Cloning
The initial ACTB cDNA fragment isolated from little skate liver was cloned using the Invitrogen Gateway pENTR-D-TOPO entry vector kit. The sequence was ligated into the pENTR vector directly from the PCR reaction to increase the efficiency of the cloning process. TOPO One Shot Competent E.coli MACH1 cells were used to propagate the pENTR_LeACTB_LI-1 vector. Cells were plated on luria broth (LB) agar plates containing 50 μg/ul Kanamyacin. Colonies with the ACTB insert were selected for by the kanamyacin resistance gene included within the pENTR vector. Clones selected for further analysis were purified from 2 ml cultures by precipitation with 2.5 M ammonium acetate, 100% isopropanol and 70% ethanol. The purified plasmid DNA was subjected to a double restriction enzyme digest with KpnI and ApaI to screen for the correct size of the insert. Digested DNA was analyzed by electrophoresis on a 1.2% agarose gel to separate the fragments. A liver sample which contained the appropriate sized fragments was selected for further purification by the Qiagen Miniprep DNA purification kit following manufacturer's instruction and sequenced using the pUC universal M13 forward and M13 reverse sequencing primers, which produce nearly complete overlapping sequences of the entire cDNA insert. Multiple sequencing reactions were prepared to check the direction of the ACTB sequence in the vector. The vector containing the initial fragment of ACTB (from liver) was used in the construction of the qPCR standard curve.
A total of 3 μl of the 250 base pair 5' RACE product and the 700 base pair 3' RACE PCR product were directly added into the corresponding ligation reaction into Promega pGEM-T Easy vectors. Competent JM109 cells were heat-shock transformed with the recombinant plasmids containing the RACE ACTB inserts. The cells were plated on LB/ agar plates containing 50 μg/ul, ampicillin, with 100 μl 1M Isopropyl β-D-1-thiogalactopyranoside (IPTG) and 20 μl Xgal applied to the surface to facilitate identification of sequence containing clones (blue white colony selection). Clones selected for further analysis were grown in an additional 2 ml LB broth with 50 μg/ul, ampicillin. The plasmids were purified and digested with the restriction enzyme EcoRI. Digests were analyzed by electrophoresis on a 1% agarose gel as described above to separate the fragments. Two clones from each RACE were chosen and the plasmids were purified using the Qiagen Miniprep DNA purification kit. The samples were then prepared for sequencing using the pUC universal M13 forward and M13 reverse sequencing primers.
Sequencing
Sequencing reactions were performed by the Rhode Island Genomics and Sequencing Center located at the University of Rhode Island. Sequencing results were aligned to form one contiguous ACTB cDNA sequence using the Sequencher program (Gene Codes Corporation, Ann Arbor, MI).
Real-time PCR
A total reaction volume of 10 μl was prepared for each qPCR using a final concentration of 1X SYBR Green Supermix (BioRad, Hercules, CA), 0.5 μM of each forward and reverse ACTB primer and 1 μl of pENTR_LeACTB plasmid. All reactions were performed in 96-well plates in triplicate reactions with optimized cycling parameters and a melt curve analysis (Table 1) using a iQ5 Imaging Module and iCycler (BioRad).
Results
For this experiment, we successfully cloned and sequenced the little skate ACTB gene. The aligned contiguous cDNA of the initial liver sequence and the 3' and 5' pancreas RACE products resulted in 1722 nucleotides including 80 base pairs of 5' untranslated region (UTR) and 510 base pairs of 3' UTR through the poly-adenylated tail. The total resulting ACTB coding region is 1127 nucleotides in length, producing a deduced amino acid sequence 374 amino acids in length with a predicted mass of 41.7 kDa (Figure 1). The complete cDNA sequence was deposited into NCBI GenBank (accession EU701009).

Figure 1. Leucoraja erinacea ACTB cDNA sequence and deduced amino acid sequence (GenBank Accession No. EU701009)
A clustal W multiple sequence alignment of the deduced little skate ACTB amino acid sequence to a number of other vertebrates shows the conservation of the protein (Figure 2).

Figure 2. A clustal W amino acid sequence alignment of the deduced ACTB sequence of little skate, Leucoraja erinacea (Leuer), GenBank Accession No. EU701009, zebrafish, Danio rerio (Danre) AAH45879, human, Homo sapiens (Homsa) BAD96645, leopard shark, Triakis semifasciata (Trisc) BAB1355, chicken, Gallus gallus (Galga) CAA25004, western clawed frog, Xenopus tropicalis (Xentr) NP_989332, Florida lancelet, Brachiostoma floridae (Brafl) Q93131, and yeast, Saccharomyces cerevisiae (Sacce) IYVN_A.
The little skate ACTB contains the conserved ATP, gesolin and profilin binding sites found on human ACTB (Figure 3). Little skate ACTB exhibits 99.7% homology with human ACTB (1 amino acid difference) as well as 98.9% homology with the zebrafish ortholog (4 amino acid differences) (Table 2). A clustal W multiple sequence alignment of little skate ACTB cDNA demonstrated conservation at the nucleotide level. Little skate exhibited 82% homology with the human ortholog of ACTB and 84% homology with the zebrafish ortholog.

Table 2. ACTB Sequence Percent Similarity Matrix of little skate, Leucoraja erinacea (Leuer), GenBank Accession No. EU701009, zebrafish, Danio rerio (Danre) AAH45879, human, Homo sapiens (Homsa) BAD96645, leopard shark, Triakis semifasciata (Trisc) BAB1355, chicken, Gallus gallus (Galga) CAA25004, western clawed frog, Xenopus tropicalis (Xentr) NP_989332, Florida lancelet, Brachiostoma floridae (Brafl) Q93131, and yeast, Saccharomyces cerevisiae (Sacce) IYVN_A.
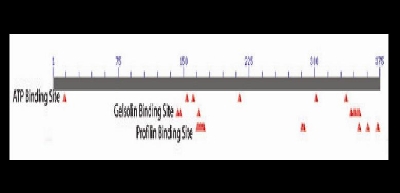
Figure 3. Diagram of conserved domains of the amino acid sequence of ACTB generated using the CDD informatics (Marchler-Bauer et al. 2007). These include the ATP binding site, the gelsolin binding site, and the profilin biding site.

Figure 4. qPCR amplification data, melt curve analysis and constructed standard curve for little skate ACTB. A. The tight grouping of the triplicate amplification lines of each of the ACTB standards shows the precision of the reactions; B. A single peak in the melt curve analysis demonstrates that one specific product was amplified; C. The standard curve generated has an efficiency value of 94.7% indicating a robust and reproducible assay for use in future research.
A standard curve using the cloned sequence of ACTB generated by qPCR resulted with an r-square value of 0.998. A standard curve using the cloned sequence of ACTB generated by qPCR resulted with an r-square value of 0.998. The efficiency of the reaction was 94.7%, which indicates close to 2 copies of the target molecule were replicated per cycle. Melt curve analysis of the PCR reaction resulted in a single peak indicating one specific product was amplified (Figure 4).
Discussion
The ACTB amino acid sequence is highly conserved among vertebrates as shown in Figure 2 and Table 2. The important functional domains in human ACTB are also conserved in little skate ACTB as shown in Figure 3 (Marchler-Bauer et al. 2007). These results were expected considering the presence and importance of the protein ACTB in nearly every cell.
For this reason, ACTB was chosen as a first pass reference gene for qPCR in little skates. Previous studies have questioned the use of ACTB as a reference gene due to changes in expression with different experimental conditions (Dheda et al. 2005). Normalization is essential to control for experimental error among samples and differences in the amount of starting material added to the qPCR. The selection of an improper reference gene that is not constitutively expressed can lead to incorrect or biased results. For example, if the experimental treatment reduced or increased the expression of the housekeeping gene, the normalized target gene quantity would be inflated or underestimated, respectively.
Our high efficiency and r-square values from the standard curve support that our experimental optimization of ACTB is a promising choice for a reference gene in future reactions. ACTB is also used as a reference gene in recent studies pertaining to determination of gene expression in fish species after exposure to toxic pollutants (Handley-Goldstone et al. 2005; Kim et al. 2008). However, further analysis of other commonly used reference genes, such as elongation factor 1 alpha, is needed to determine the optimal gene for normalization in a qPCR assay. Once the best reference gene is chosen according to the specific experimental conditions, a specific, reproducible and accurate qPCR can be performed and analyzed. We have submitted the little skate ACTB sequence to Genbank to aid other research projects using little skate. Our contribution of new genomic data for little skate broadens the use of this emerging bioindicator in ecotoxicology research and other related fields.
Acknowledgements
We thank Drs. Michael Moore, Maria Johnsson, and Mark Hahn from the Woods Hole Oceanographic institution for kindly providing tissue samples of skates from Boston Harbor. This project was made possible by RI-INBRE Grant #P20RR016457 subproject grant to RRM from NCRR, NIH, and the Bioinformatics Core at URI. Sequencing was subsidized by Rhode Island Genomics and Sequencing Center which is supported in part by the National Science Foundation (MRI Grant No. DBI-0215393 and EPSCoR Grant No. 0554548), the US Department of Agriculture (Grant Nos. 2002-34438-12688 and 2003-34438-13111), and the University of Rhode Island.
References
Ballatori, N et al (2003) Exploiting genome data to understand the function, regulation, and evolutionary origins of toxicologically relevant genes. Environmental Health Perspectives. 111: 871-875.
Bustin, S. (2005) Real-time PCR. Encyclopedia of Diagnostic Genomics And Proteomics 10: 1117-1125.
Dheda, K et al. (2005) The implications of using an inappropriate reference gene for real-time reverse transcription PCR data normalization. Analytical Biochemistry 344: 141-143.
Kim, J-H et al. (2008) Molecular cloning and B-naphthoflavone-induced expression of a cytochrome P450 1A (CYP1A) gene from an anadromous river pufferfish, Takifugu obscures. Marine Pollution Bulletin.
Koob, T and I Callard (1999) Reproductive endocrinology of female elasmobranchs: Lessons from the little skate (Raja erinacea) and spiny dogfish (Squalus acanthias). Journal of Experimental Zoology Part A: Comparative Experimental Biology 284: 557 574.
Handley-Goldstone, H et al. (2005) Cardiovascular gene expression profiles of dioxin exposure in zebrafish embryos. Toxicological Sciences 85: 683-693.
Ng, S et al (1985) Evolution of the functional human beta-actin gene and its multi-pseudogene family: conservation of the noncoding regions and chromosomal dispersion of pseudogenes. Molecular Cellular Biology. 5: 2720-2732.
NOAA Northeast Fisheries Science Center (2006) 43rd Northeast Regional Stock Assessment Workshop (43rd SAW) 43rd SAW Assessment Report. Northeast Fish Sci Cent Ref Doc 06-25.
Marchler-Bauer, A et al (2007), CDD: a conserved domain database for interactive domain family analysis. Nucleic Acids Research 35: 237-240.
Maglott, D et al (2005) Entrez Gene: gene-centered information at NCBI. Nucleic Acids Research 33: D54D58.
Mattingly, C et al. (2004) Cell and molecular biology of marine elasmobranches: Squalus acanthias and Raja erinacea. Zebrafish 1: 111-120.
Schmale, M et al (2006) Aquatic Animal Models of Human Disease. Comparative Biochemistry and Physiology, Toxicology and Pharmacology 145: 1-4.