

Author: Weisha Zhu
Institution: Cornell University
Date: September 2007
ABSTRACT
The functional properties of proteins depend on their three-dimensional shapes. Protein structures can be determined by X-ray crystallography as a tool. The three-dimensional structure of the apo form of the Escherichia coli L-arabinose isomerase (ECAI) has recently been determined. ECAI is responsible for the initial stage of L-arabinose catabolism, converting arabinose into ribulose in vivo. This enzyme also plays a crucial role in catalyzing the conversion of galactose into tagatose (low calorie natural sugar) in vitro. ECAI utilizes Mn2+ for its catalytic activity. Crystals of the ECAI + Mn2+ complex helps to investigate the catalytic properties of the enzyme. Therefore, crystals of ECAI + Mn2+ complex were grown using hanging drop vapor diffusion method at room temperature. Diffraction data were collected at X4C beamline, National Synchrotron Light Source, Brookhaven National Laboratory. The structure was solved by the molecular replacement technique and has been refined to Rwork of 0.23 at 2.8 Å resolution using X3A beamline computational facility. The structure was deposited to Protein Data Bank (PDB ID 2HXG). Mn2+ ion was localized to the previously identified putative active site with octahedral coordination. Comparison of apo and holo form of ECAI structures permits the identification of structural features that are of importance to the intrinsic activity and heat stability of AI.
INTRODUCTION
Structural genomics (SG) is one of the major branches of proteomics research and represents an international effort to determine the three-dimensional structures of proteins and other important biological molecules encoded by the genomes of key organisms (Burley 2000; Chance et al. 2002; Heinemann 2000; Stevens et al. 2001). There are a number of groups funded around the globe to obtain a large number of protein structures using technologies such as X-ray crystallography and nuclear magnetic resonance. In USA, the New York Structural GenomiX Research Consortium (NYSGXRC) is one of the large scale centers for the Protein Structure Initiative (PSI) funded by National Institute of Health that adopts high throughput techniques to solve structures across all 5000 protein families (Bonanno et al. 2005; Manjasetty et al. 2007). The project outlined in this article analyzes structure-to-function analysis of one of the many selected targets chosen by NYSGXRC (T2031).
Arabinose can be utilized as a sole carbon source by many microorganisms. This utility is accomplished via the pentose-phosphate pathway converting arabinose into ribulose, in vivo. An intermediate in the pathway, D-xylulose-5-phosphate, is converted from L-arabinose in three steps under regulation of distinct enzymes (Lee et al. 1986). In Escherichia coli, the enzyme L-arabinose isomerase (ECAI, EC 5.3.1.4; swiss-prot P08202; Mr = 56,043 Da; 500 amino acid residues) permits the conversion of L-arabinose to L-ribulose in vivo. This enzyme is unusual as it is able to isomerize structurally related sugars in vitro. Most notably, the enzyme carries out the isomerization of D-galactose to D-tagatose (Scheme 1) with a conversion yield of 34% at reaction temperature of 35˚C (Roh et al. 2000). Its alternative name, D-galactose isomerase, is derived from this function.

article_1255_order_0
The compound tagatose is a rare natural sugar and reported as a sweetener that has similar taste and properties as sucrose but fewer calories (Levin et al. 1995). The industrial demand for high production of low-calorie sugar by enzymatic method calls for cost effective galactose-tagatose isomerization reaction (Jorgensen et al. 2004). The insertion of arabinose isomerase (AI) to mediate this reaction is more efficient compared to other methods such as tagatose production using several micro-organisms by oxidation of galacitol(Izumori and Tsuzaki 1988; Izumuri et al. 1984) or when galactose derived from lactose hydrolysis is isomerized by a calcium catalyst (Beadle et al. 1992). In view of the enzyme's potential industrial utility and to better understand the reaction mechanism, further structural knowledge of AI is crucial. We have recently presented the crystal structure of apo form of ECAI (Manjasetty and Chance 2006). The apo model confirmed many speculations from previous thermodynamic and biophysical examinations on ECAI and mapped out potential metal-binding activation sites on the structural model. The majority of L-arabinose isomerases are metalloproteins involving a relatively high concentration (1 to 5mM) of metallic ions for their optimal activity and thermostability (Rhimi and Bejar 2006). Specifically, in the case of ECAI, Mn2+ binding produces an enzyme with greater intrinsic activity and heat stability (Banerjee et al. 1995; Patrick et al. 1971).
To continue investigating the role of divalent cations in the structure of ECAI, as well as to further study the biological properties of the enzyme, we have co-crystallized ECAI with a metal cofactor (Mn2+) and candidate substrates (L-arabinose and D-galactose) / inhibitors (ribitol and xylitol). Here, we report the direct evidence for the binding of metal ions to the putative active site of ECAI and the crystal structure of Mn2+ bound ECAI protein complex was solved and refined to 2.8 a Å resolution.
MATERIALS AND METHODS
ECAI Purification, Complexes and Crystallization
The protein of ECAI was obtained from SGX Pharmaceuticals Inc., San Diego, CA (Target T2031). The protein was expressed in E. coli and purified by standard approaches (Bonanno et al. 2005). Briefly, a full-length cDNA fragment of L-arabinose isomerase (GenBank BAB96631) was amplified by Polymerase Chain Reaction (PCR) from E. coli genomic DNA using forward and reverse primers. The PCR product was cloned into a pET vector modified for topoisomerase directed cloning (Invitrogen) designed to express the protein of interest followed by a C-terminal hexa-histidine tag; TOP10 cells were transferred with this vector. The resulting clone was grown by adding 500mL of Luria-Bertani (LB) medium containing 500µL of 30mg/mL kanamycin, 25mL of 10% glucose, and a small amount of transformed cell glycerol stock scraping to a 2L baffled flask at 30°C with overnight shaking (250rpm). A 10mL of the resulting culture was added to each of six flasks containing similar culture medium for large-scale expression. The cultures were subjected to shaking under similar conditions until the OD595 reached to the range of ~0.8. The protein expression was induced by adding 200µL of 1M isopropyl-D-thiogalactopyronoside (IPTG). After overnight vigorous shaking (250rpm, 21°C), the cells were pelleted by centrifugation (in 1L spin bottles; at 6500rpm for 10 min). The pellets were then collected into 50mL conical tubes and re-suspended in a lysis buffer (35mL/10g), 50 µL of protease inhibitor cocktail tablet (Sigma) and 5 µL of benzonase (Novagen). The cells were lysed by repeated sonication (with intervals of cooling) followed by centrifugation (38,900 g for 30 min). The filtrate was then immobilized on Ni-NTA-agarose resin (Qiagen), placed on a drip column and washed with 25mL buffer-A (50mM Tris-HCl pH7.8, 500mM NaCl, 10mM imidazole, 10mM methionine, and 10%glycerol). The protein was eluted into Amicon concentrator (Millipore) with 15mL of buffer-A containing 500mM imidazole. The eluted protein was then concentrated to 6 mL and loaded onto gel filtration (Superdex 200, Pharmacia) column. The fractions were pooled, and protein was concentrated to 18.6mg/ml and stored in 10mM Hepes pH7.5, 150mM NaCl, 10mM methionine, 10% glycerol.
Complexes of ECAI with and without metal cofactor (Mn2+) and candidate substrates (L-arabinose & D-galactose) / inhibitors (ribitol & xylitol) were prepared. Diffraction quality crystals were grown at room temperature by the hanging drop vapor diffusion method (Durbin and Feher 1996; Luft and DeTitta 1992). The droplets consisted of 1.4 μL of protein complex plus 1.4 μL reservoir solution (18% polyethylene glycol 3350 and 0.2 M tri-sodium citrate dehydrate) equilibrated against 750 μL of reservoir solution. Crystals suitable for X-ray diffraction were allowed to form for a two day period.
Data Collection and Processing
X-ray data sets of ECAI complex crystals were collected at 100K using the X29, X4C and X6A beamlines of National Synchrotron Light Source (NSLS), Brookhaven National Laboratory (BNL). A solution of 20% glycerol in the crystallization solution was used as cryoprotectant. Diffraction data were integrated and scaled with program HKL2000 (Otwinowski and Minor 1997). The collected data were truncated using TRUNCATE of CCP4 programs (Collaborative Computational Project Number 4 1994). Data collection statistics are given in Table 1. The crystallographic data collected on ECAI + Mn2+ complex was used for further analysis. All additional computation was performed using X3A beamline computational facility.

article_1255_order_1
Structure Solution and Refinement
Although the unit cell parameters and space group are quite similar to the ECAI apo structure, the straight refinement with the program REFMAC5 (Murshudov et al. 1997) yielded an elevated Rfree and Rwork values. Therefore, the structure of ECAI metal complex were solved by molecular replacement using the program MolRep (Vagin and Teplyakov 2000). The crystal structure of ECAI (PDB code 2AJT) was used as the starting model. The rotation and translation search yielded three peaks corresponding to three molecules in the asymmetric unit with a correlation coefficient of 0.659 and an R value of 45.1%. Density improvement and removal of model bias was achieved by the free-atom refinement method in ARP/wARP (Perrakis et al. 2001). The model rebuilding was performed using the program COOT (Emsley and Cowtan 2004). The model was refined using the program REFMAC5 (Murshudov et al. 1997) with the TLS parameters (Winn et al. 2001). 2Fo-Fc and difference Fourier maps were clearly shown for electron-density corresponding to bound Mn2+ ion. The final model includes 1494 residues; 3 Mn2+ ions and 106 water molecules. The refinement statistics are shown in Table 2. The coordinates and structure factors have been deposited with the Protein Data Bank with the accession code 2HXG. Figures were generated using the program PyMol (DeLano 2003).

article_1255_order_2
RESULTS
Overall Structure
The crystals of ECAI grown in the presence of Mn2+ ions are shown in Figure 1A. X-ray diffraction data (as seen in Figure 1B) collected to 2.8 Å resolution. The overall fold of the monomer (and domains) are retained in the Mn2+ bound structure of ECAI (Figure 1C) similar to the apo structure of ECAI(Manjasetty and Chance 2006) with root mean square (r.m.s.) deviation of 0.36 Å. The asymmetric unit contains ECAI trimer (Figure 1D) and the functional hexamer (336-kDa) is generated by crystallographic dyad symmetry (Figures 1E & F).
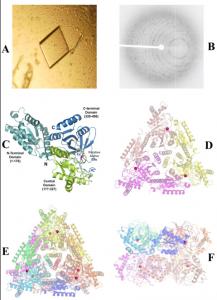
article_1255_order_3

article_1255_order_4
Cation binding site
The active center location of ECAI is shown in Figure 1C. The electron density within the active site indicates the presence of a tightly bound metal ion (Figure 2A). Since crystals were grown in the presence of 2mM MnCl2, the metal ion bound to the enzyme in the crystal structure has been assigned as Mn2+. The Mn2+ ion ligands to the residues His350, His450, Glu303, Glu333 and two water molecules (wat 2 and wat 3) which completes the octahedral coordination.
Substrate binding pocket
The substrate binding pocket was located at the adjacent subunit interface as seen in Figure 2D. The pocket reaches about 20Å below the molecular surface. The entrance of the pocket is closed and filled with one water molecule (wat59) as seen in Figures 2A and 2E. The pocket is surrounded by central and C-terminal residues of one subunit and residues from the N-terminal domain of the neighboring subunit. The majority of these residues lining the pocket are located at the loop regions (Figure 2E).
Comparison of ECAI structures
Despite the overall structural similarity between apo and holo structures of ECAI, the two structures show major variations at the putative active site region and at the substrate binding pocket entrance. Firstly, the Mn2+ ion is bound at a substrate binding pocket in the holo structure in an exactly similar manner as water bound (Wat20) in the apo structure (Figure 2A and 2B). However, the geometry of the active site quite different between the two structures. Data on the active site ligands for both structures are given in the table 3; an average ligand distance of 2.63 Å and 2.26Å for apo and holo structures is observed, respectively. The active site residue Glu306 has different conformations between the two structures as shown in Figure 2C. A major structural rearrangement is observed at the residues from loop regions from all three domains as shown in Figure 2F.

article_1255_order_5
DISCUSSION AND CONCLUSION
Industrial Importance and Catalytic Mechanism
D-tagatose, an isomer of D-galactose, is a novel natural ketohexase having a taste and physical properties similar to sucrose (Levin 2002; Levin et al. 1995). Therefore, AI is of industrial importance because of its action as a galactose isomerase catalyzing the conversion of D-galactose to the novel sweetner, D-tagatose (Kim 2004). The catalytic action has been reported to involve metal-mediated proton transfer converting an aldose to a ketose through an ene-diol intermediate (Banerjee et al. 1995). Based on the previous crystal structure of ECAI, it can be seen that the hydroxyl groups of Glu306/Glu333 act as the general base within the ene-diol mechanism. Interestingly, in the Mn2+ bound structure, the residue Glu306 acquired a different conformation than in the apo structure suggesting its importance to metal binding (Figure 2C). A comparison of active site geometry of apo and holo structures suggests the Mn2+ bound structure forms a more stable active site (table 3). These structural observations might be one of the factor contributing to greater catalytic activity in the presence of Mn2+ (Banerjee et al. 1995; Lee et al. 2005; Patrick et al. 1971).
Thermostability
Biochemical studies also suggest that ECAI in the presence of Mn2+ ion at the active site will exhibits greater heat stability than the apo form. Several structural features have been suggested to contribute to the stability of the proteins. These features include the increase in hydrogen bonds and ion pairs (Vogt and Argos 1997; Vogt et al. 1997) and a decrease in accessible surface area (Chan et al. 1995). The structure of Mn2+ bound ECAI forms an active site with completed octahedral coordination and the entrance of the substrate binding pocket is closed (not solvent accessible) as shown in Figure 2D. The major structural rearrangements at the entrance of the substrate binding pocket leads to an increasing number of hydrophobic interactions at the subunit interface (Figure 2F). In addition, the substrate binding pocket is filled with water molecules and the entrance is closed as seen is Figure 2E. The decrease in solvent accessible surface area and increase in hydrophobic interactions between the subunits are the most probable structural factors which lead to the greater heat stability in the presence of Mn2+.
High-throughput techniques for protein structure analysis were also adopted in structure elucidation. The analysis of the structure of Mn2+ bound ECAI protein complex completed to 2.8 Å resolution with the R-factor of 22.9%. Comparison between the native and complex crystal forms reveals variations in the metal coordination sphere and conformational changes associated with the active site residues. Previous biochemical and present structural data were used to explain the possible reasons for greater intrinsic activity and heat stability of the enzyme induced by the binding of Mn2+ at the putative active site. This study will benefit optimization of future low-calorie sugar production.
ACKNOWLEDGEMENTS
The author wishes to thank the US Department of Energy for the Science Undergraduate Laboratory Internship (SULI) and Mike Sullivan, John Toomey, Donald Abel of Case Center for Synchrotron Biosciences, National Synchrotron Light Source (NSLS) at Brookhaven National Laboratory (BNL). We thank the X3A, X4C, X6A and X29 beamline staffs at NSLS and the Office of Educational Program at BNL for their generous support. This research is supported by the Biomedical Technology Resource Program of the National Institute for Biomedical Imaging and Bioengineering under P41-EB-01979 and the Protein Structure Initiative funded by the National Institute for General Medical Sciences under U54-GM-74945.
REFERENCES
Banerjee, S., Anderson, F. and Farber, G.K. (1995) The evolution of sugar isomerases. Protein Eng, 8, 1189-1195.
Beadle, J.R., Saunder, J.P. and Wajada, T.J. (1992) Process of manufacturing tagatose. US Patent 5,078,796.
Bonanno, J.B., Almo, S.C., Bresnick, A., Chance, M.R., Fiser, A., Swaminathan, S., Jiang, J., Studier, F.W., Shapiro, L., Lima, C.D., Gaasterland, T.M., Sali, A., Bain, K., Feil, I., Gao, X., Lorimer, D., Ramos, A., Sauder, J.M., Wasserman, S.R., Emtage, S., D'Amico, K.L. and Burley, S.K. (2005) New York-Structural GenomiX Research Consortium (NYSGXRC): A Large Scale Center for the Protein Structure Initiative. J Struct Funct Genomics, 6, 225-232.
Burley, S.K. (2000) An overview of structural genomics. Nat Struct Biol, 7 Suppl, 932-934.
Chan, M.K., Mukund, S., Kletzin, A., Adams, M.W. and Rees, D.C. (1995) Structure of a hyperthermophilic tungstopterin enzyme, aldehyde ferredoxin oxidoreductase. Science, 267, 1463-1469.
Chance, M.R., Bresnick, A.R., Burley, S.K., Jiang, J.S., Lima, C.D., Sali, A., Almo, S.C., Bonanno, J.B., Buglino, J.A., Boulton, S., Chen, H., Eswar, N., He, G., Huang, R., Ilyin, V., McMahan, L., Pieper, U., Ray, S., Vidal, M. and Wang, L.K. (2002) Structural genomics: a pipeline for providing structures for the biologist. Protein Sci, 11, 723-738.
Collaborative Computational Project Number 4. (1994) The CCP4 suite: programs for protein crystallography. Acta. Cryst., D50, 760-763.
DeLano, W.L. (2003) The PyMOL Molecular Graphics System. DeLano Scientific, San Carlos, CA, USA http://www.pymol.org.
Durbin, S.D. and Feher, G. (1996) Protein crystallization. Annu Rev Phys Chem, 47, 171-204.
Emsley, P. and Cowtan, K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr, 60, 2126-2132.
Heinemann, U. (2000) Structural genomics in Europe: slow start, strong finish? Nat Struct Biol, 7 Suppl, 940-942.
Izumori, K. and Tsuzaki, K. (1988) Production of D-tagatose from D-galacitol by Mycobacterium smegmatts. J.Ferment.Technol., 66, 225-227.
Izumuri, K., Miyoshi, T., Tokuda, S. and Yamabe, K. (1984) Production of D-tagatose from ducitol by Arthrobacter globiformis. Appl.Environ.Microbiol., 46, 1055-1057.
Jorgensen, F., Hansen, O.C. and Stougaard, P. (2004) Enzymatic conversion of D-galactose to D-tagatose: heterologous expression and characterisation of a thermostable L-arabinose isomerase from Thermoanaerobacter mathranii. Appl Microbiol Biotechnol, 64, 816-822.
Kim, P. (2004) Current studies on biological tagatose production using L-arabinose isomerase: a review and future perspective. Appl Microbiol Biotechnol, 65, 243-249.
Lee, D.W., Choe, E.A., Kim, S.B., Eom, S.H., Hong, Y.H., Lee, S.J., Lee, H.S., Lee, D.Y. and Pyun, Y.R. (2005) Distinct metal dependence for catalytic and structural functions in the L-arabinose isomerases from the mesophilic Bacillus halodurans and the thermophilic Geobacillus stearothermophilus. Arch Biochem Biophys, 434, 333-343.
Lee, N., Gielow, W., Martin, R., Hamilton, E. and Fowler, A. (1986) The organization of the araBAD operon of Escherichia coli. Gene, 47, 231-244.
Levin, G.V. (2002) Tagatose, the new GRAS sweetener and health product. J Med Food, 5, 23-36.
Levin, G.V., Zehner, L.R., Saunders, J.P. and Beadle, J.R. (1995) Sugar substitutes: their energy values, bulk characteristics, and potential health benefits. Am J Clin Nutr, 62, 1161S-1168S.
Luft, J.R. and DeTitta, G.T. (1992) HANGMAN: A macromolecular hanging drop vapor diffusion technique. J. App. Cryst., 25, 324-325.
Manjasetty, B.A. and Chance, M.R. (2006) Crystal structure of Escherichia coli L-arabinose isomerase (ECAI), the putative target of biological tagatose production. J Mol Biol, 360, 297-309.
Manjasetty, B.A., Shi, W., Zhan, C., Fiser, A. and Chance, M.R. (2007) A high-throughput approach to protein structure analysis. Genet Eng (N Y), 28, 105-128.
Murshudov, G.N., Vagin, A.A. and Dodson, E.J. (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallographica Section D-Biological Crystallography, V53, 240-255.
Otwinowski, Z. and Minor, W. (1997) Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol., 276, 307-326.
Patrick, J.W., Lee, N., Barnes, N.B. and Englesberg, E. (1971) Coordination of enzyme synthesis in the L-arabinose operon in Escherichia coli. I. The effect of manganous ion on the synthesis of L-arabinose isomerase. J Biol Chem, 246, 5102-5106.
Perrakis, A., Harkiolaki, M., Wilson, K.S. and Lamzin, V.S. (2001) ARP/wARP and molecular replacement. Acta Crystallogr D Biol Crystallogr, 57, 1445-1450.
Rhimi, M. and Bejar, S. (2006) Cloning, purification and biochemical characterization of metallic-ions independent and thermoactive l-arabinose isomerase from the Bacillus stearothermophilus US100 strain. Biochim Biophys Acta, 1760, 191-199.
Roh, H.J., Kim, P., Park, Y.C. and Choi, J.H. (2000) Bioconversion of D-galactose into D-tagatose by expression of L-arabinose isomerase. Biotechnol Appl Biochem, 31 ( Pt 1), 1-4.
Stevens, R.C., Yokoyama, S. and Wilson, I.A. (2001) Global efforts in structural genomics. Science, 294, 89-92.
Vagin, A. and Teplyakov, A. (2000) An approach to multi-copy search in molecular replacement. Acta Crystallogr D Biol Crystallogr, 56 Pt 12, 1622-1624.
Vogt, G. and Argos, P. (1997) Protein thermal stability: hydrogen bonds or internal packing? Fold Des, 2, S40-46.
Vogt, G., Woell, S. and Argos, P. (1997) Protein thermal stability, hydrogen bonds, and ion pairs. J Mol Biol, 269, 631-643.
Winn, M.D., Isupov, M.N. and Murshudov, G.N. (2001) Use of TLS parameters to model anisotropic displacements in macromolecular refinement. Acta Crystallogr D Biol Crystallogr, 57, 122-133.