

Author: Marina Freydin
Institution: University of Maryland
Date: October 2007
ABSTRACT
Hemophilias are genetic bleeding disorders for which there are still no cures. Treatment of hemophilias are difficult because patients need repeated infusion of missing coagulation factors, some patients develop inhibitors to the infused factors, and gene therapy is still not suitable for mass treatment. The choice between the two most common therapies, plasma-derived products or recombinant factor VIII or factor IX, is still a dilemma for clinicians involved in the care of patients with hemophilia. This review article details three primary types of treatment methods for patients with hemophilias A and B: plasma-derived products, recombinant factors, and gene therapy, as well as the therapies for patients who develop inhibitors: activated Prothrombin complex concentrates, recombinant factor VIIa, and immunosuppressive or immuno-tolerance inducing treatments. The review also examines the available evidence in relative effectiveness of the different treatment options available in hemophilia patients who do and do not develop inhibitors to treatment. Ultimately, it is suggested that recombinant factors are a preferred treatment method for patients free of inhibitors and FVIIa treatments are the suggested method for patients that do develop inhibitors to the aforementioned regimen.
INTRODUCTION
Hemophilia is genetic disorder that affects over 18,000 people (most of whom are males) in the United States alone (National Heart, Blood, and Lung Institute 2006). Hemophilia is caused by a deficiency in a gene on the X chromosome coding for a specific coagulating factor (National Heart, Blood, and Lung Institute 2006) or the development of antibodies that destroy coagulating factors (Green 2006).
The most common form of hemophilia, hemophilia type A (classic hemophilia), is caused by an insufficiency in clotting factor VIII and the second-most common form of the disease, hemophilia type B (Christmas hemophilia), is caused by an insufficiency in clotting factor IX (Ingram 1976). Both hemophilia A and B are present at birth and affected children must receive immediate treatment. In the United States, 400 babies per year are born with the disease and without proper treatment, most will not survive to adolescence (National Heart, Blood, and Lung Institute 2006). Acquired hemophilia, however, results from an autoimmune attack against clotting factors, usually factor VIII, and occurs later in life. This disease is rare and is generally linked with cancer and certain autoimmune diseases (Green 2006).
The process of blood coagulation occurs through a series of activating steps whereby one enzyme activates another enzyme that then activates another enzyme and so on (figure 1). Understanding the sequence of coagulation assists in deducing the correct treatment options for patients with a missing link in the sequence. Coagulating factors are produced in the liver by hepatocytes. The end products of the coagulation cascade are polymers of fibrin, which prevent blood loss upon injury by associating with aggregates of platelet cells.
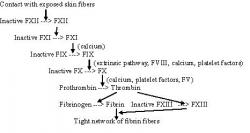
Figure 1 - Blood Coagulation Cascade: In the blood coagulation cascade factors VIII and IX play an important role in clot formation. The activated factors stimulate the conversion of other factors into their active state and so on, until fibrin fibers form a tight meshwork. Items marked by parentheses are also responsible for the activation step of indicated factors. Figure adapted from Germann and Stanfield (2005).
Symptoms of hemophilia can range from mild to severe depending on the amount of clotting factors present in blood. In general, patients with hemophilia bleed for a longer period of time than healthy individuals. Common symptoms include large bruises, spontaneous bleeding from gums or nose, tightness in joints, and blood in emesis or feces. Severe symptoms of hemophilia include bleeding in the joints, brain, or internal organs; profuse bleeding after injury or surgery; and anemia, which can lead to shock and death. Bleeding in the joints can develop into swelling that, over time, breaks down cartilage and bone and can progress to chronic joint pain and immobility (National Heart, Blood, and Lung Institute 2006). Acquired hemophilia presents with distinct symptoms such as bleeding into muscle and soft tissue and into the gastrointestinal and urinary tracts (Green 2006).
The types of treatment methods available today are plasma-derived products, recombinant coagulating factors, and gene therapy. Patients who develop an immune response to therapy or have acquired hemophilia are extremely difficult to treat (Mannucci 2003). The treatment options for these patients include prothrombin complex concentrates, activated recombinant factor VII therapies, and immunosuppressive drugs.
The most common hindrance in hemophilia treatment is the production of inhibitors, or antibodies against injected replacement coagulating factor. High (2006) notes that as many as 20% of hemophilia patients develop inhibitors to treatment. Acquired hemophilia patients also have antibodies against clotting factors and thus, the treatment methods for both of these patient groups are similar. Although hemophilia treatments are advanced and readily available in developed countries, it is estimated that 70% of the people with this disease worldwide are undiagnosed or under-treated (O'Mahony and Black 2005). Reasons for poor treatment include lack of knowledge about the disease, the presumption that hemophilia is very rare, and inadequate access to medications (O'Mahony and Black 2005). Whether to choose plasma-derived or recombinant factor VIII or factor IX is a dilemma for clinicians involved in the care of patients with hemophilia. Safety, cost, availability are the determining factors (Manucci and Tuddenham 2001).
This review discusses treatment options for hemophilia patients without inhibitors as well as for those who develop immune response to therapy. Methods of treatment are reviewed based on their effectiveness, associated side effects, and feasibility for use.
TREATMENT OPTIONS FOR PATIENTS WITHOUT INHIBITORS
EFFECTIVENESS OF PLASMA- DERIVED PRODUCTS
Transfusion of blood, plasma-derived products, or even tissues with healthy cells has been a treatment option for hemophilia patients since the 1970s (Farrugia 2002). The coagulating factors in whole blood and in plasma fractions were used as replacements for any absent or dysfunctional factors in hemophiliacs (Ludlam et al. 2006). Plasma products currently available are fresh frozen plasma, freeze-dried concentrates, and cryoprecipitate (slowly thawed plasma precipitate) (Ingram 1976). Ingram (1976) notes that blood transfusions were the earliest most successful treatment of hemophilia. Transfusions with healthy blood not only provide the patient with missing clotting factors necessary for coagulation, but also replenish blood volume depleted during excessive bleeds. Although therapeutic benefits are present, re-injection with product is necessary as live tissue cells and cell products must be replenished over time.
The success of the treatment lies in the methods used to cleanse blood and plasma products from pathogens. After the adoption of moderate dry heating, strong dry heating, and wet heating of blood products in the 1980s, the occurrence of hemophilia patients contracting the deadliest of blood-borne pathogens, Human Immunodeficiency Virus (HIV) and Hepatitis C, has not been documented (Mannucci 2003). According to Ludlam et al. (2006), detection methods such as nucleic-acid screening and incorporation of products that reduce viral activity have made blood products safe from hepatitis B (HBV), hepatitis C (HCV), HIV, and human T-cell lymphotropic virus (HTLV). The current focus is on the use of reagents that are totally independent of human plasma (Ludlam et al. 2006).
ASSOCIATED PROBLEMS
In the 1980s a tragic crisis emerged in Europe and the United States: 60-80% of patients with severe hemophilia contracted HIV from blood-derived products (Mannucci 2003). Most of the viruses that are commonly transmitted through blood transfusions have a long, asymptomatic carrier state indicating that persons might have the virus but show no symptoms. The asymptomatic period poses a problem because seemingly healthy people who elect to give blood may actually be carriers of a pathogen. Viruses that meet this criterion are HBV, HCV, West Nile virus, HIV, and HTLV. Although standard screenings have removed blood samples that carry these viruses, there are still potential hazards to patients (Ludlam et al. 2006).
First, since viruses can evolve into different strains or different pathogens altogether, routine techniques might not be able to detect these changes in blood samples. The transfer of a new variation of a defective prion protein associated with Creutzfeldt-Jakob disease has been shown in animal models including rodents (Evatt et al. 1998) and sheep (Hunter et al. 2002). Although the Evatt et al. (1998) retroactive study found no evidence of said transmission in humans, there is still no compelling evidence to rule out the possibility of this occurring. Second, certain pathogens are resistant to current blood purification treatments. B19 is a heat-resistant parvovirus that has been transmitted in blood despite current purification methods. Third, there is potential threat of emerging viruses. Emerging viruses develop when a pathogen first makes contact with a new species or population and establishes itself in that population. Interspecies jumping is a common route for emerging viruses.
Another potential hazard is lipid-enveloped viruses, called flaviviruses, which are common to insects and animals and are resistant to current purification treatments (Ludlam et al. 2006). Dengue fever and dengue heamorrhagic fever viruses (transferred through mosquitoes) are the most common forms of flavivirus in the world. Although there have been no cases reported in the United States after the DDT insecticide spraying in the 1950s (Reiter et al. 2003), epidemics have been reported in Brazil and Central America. Mortality rates of affected individuals have been reported to be as high as 40% (Ludlam et al. 2006). Non-lipid-enveloped viruses, such as Enterovirus and Parvovirus, are even more adept at evading purification methods because they are smaller and have a greater resistance to detergents and heat. For this reason, emerging non-lipid-enveloped viruses pose a huge threat to blood products (Ludlam et al. 2006).
The possibility of developing an adverse immune reaction to the blood-based products is also a possibility. Mannucci (2003) notes that these products are associated with about the same number of cases of inhibitor formation as recombinant factors.
AVAILABILITY AND ACCESSIBILITY
Blood transfusions and other plasma-derived factors are generally cost effective (Farrugia 2002); however, proper purification techniques are generally not accessible in some third-world countries, making safe transfusions a limited possibility. Eighty percent of hemophiliacs worldwide continue to use plasma-derived factors due to unavailability of other methods (Farrugia 2002).
RECOMBINANT FACTORS
EFFECTIVENESS
Recombinant clotting factors treatments deliver clotting factors that the hemophilia patients are missing. For example, in the most common type of hemophilia, hemophilia A, patients receive factor VIII replacements (rFVIII) and in the second most common type of hemophilia, hemophilia B, patients receive factor IX replacements (rFIX) (Meng et al. 2006). Recombinant factors are derived from DNA and although albumin is used in synthetic steps, it is not included in the final product. Newer recombinant factors have been produced that use no human proteins in the synthetic or final stages of production. These factors are thought to be safer in terms of viral transmission (Mannucci 2003). However, recovery time for patients using rFIX has been shown to be slower than those for plasma-derived factors (White et al. 1998).
Eighty percent of uncontrolled bleeds have been effectively eliminated with a single dose of recombinant factors and subsequent use increases the success rate to 90-95% (Mannucci 2003). Lusher et al. (1993) treated 95 moderate to severe hemophiliac children with rFVIII for an average of 1.5 years with an average of 34.9 infusions per individual. Injections of rFVIII were given in response to excessive bleeds and prior to surgical/dental procedures. Lusher et al. (1993) reported that all patients responded well to the treatment with minor side effects experienced by 3 patients. Batorova and Martinowitz (2002) suggest a different method of administering injections. They favor continuous injection of clotting factor (at a rate that corresponds with its clearance from the body), which prevents highs and lows in coagulating factor level and promotes homeostasis, stopping large bleeds before they occur. This method also reduces the overall amount of factor required for treatment. Clearance of recombinant factors is noted to occur through the low-density lipoprotein receptor and low-density lipoprotein receptor-related protein. Admission of antagonists for these receptors may decrease recombinant factor clearance (Saenko and Ananyeva 2006).
ASSOCIATED PROBLEMS
The development of inhibitors is the major problem associated with patients who use recombinant factors. Since hemophiliacs miss a normally functional coagulating factor, the body's immune system sees an introduced factor as a foreign substance. In an effort to rid the body of unrecognized foreign matter, an immune response forms and will ultimately destroy the infused coagulating factor (Goudemand et al. 2006). Patients with inhibitors generally exhibit increased bleeding patterns that can be life threatening (Franchini et al. 2006).
Previous studies have claimed that the incidence of inhibitors among patients could be as low as 3.6% or as high as 25% (Ehrenforth et al. 1992). However, Ehrenforth and colleagues (1992) suggest that these values are underestimated. In their study, 46 patients with hemophilia A and 13 patients with hemophilia B who had previously received factor replacement therapy were monitored for the presence of inhibitors. It was found that only those patients who received FVIII replacements developed inhibitors (24% of total hemophilia A patients and 54% of severe hemophilia A patients). The overall calculated risk for developing inhibitors after replacement therapy was 33% by the age of 6. Ehrenforth et al. (1992) claim that the risk of inhibitor formation increases if there is history of previous treatment with recombinant factor infusions as well as if the disease is severe.
Knobel et al. (2002) tested 116 persons with hemophilia for presence of inhibitors after factor replacement treatment that lasted 14-16 days. Of these people, 19% of hemophilia A patients developed inhibitors compared to 37% of hemophilia B patients. The study also suggests that development of inhibitors is characterized by genotype since all subjects who developed inhibitors had impaired protein synthesis due to a genetic mutation.
AVAILABILITY AND ACCESSIBILITY
Recombinant factors cost 20-50% more than plasma-based factors. Nonetheless, these factors are used by 60-70% of severe hemophiliacs in America and all patients in Canada and Ireland use recombinant factors over plasma-based products. Increased cost of recombinant factor product may be because the amount of coagulating factor that can be extracted from a blood sample is only 5-10% of the quantity of factor present in the sample (Mannucci 2003). Additionally, because factor FVIII is a large (280kD), complex protein product, it is difficult to synthesize (VandenDriessche et al. 2001). The discrepancy in price makes recombinant factors unaffordable for patients in developing countries (Farrugia 2002). Methods such as the use of antagonists for factor clearance receptors (Saenko and Ananyeva 2006), continuous infusion of product (Batorova and Martinowitz 2002), and increased half-life of recombinant factor (Mannucci 2003) may decrease the amount of factor needed and thus defray costs.
The presence of inhibitors after treatment is the most significant problem facing patients. Although patients may adjust the dose of the product, there is still no complete resolution of this problem (Lusher et al. 1993). Possible solutions include re-engineering the recombinant factor so that it is less likely to illicit an immune response in patients (Barrow et al. 2000).
ALTERNATE TREATMENTS FOR PERSONS WHO DEVELOP INHIBITORS
Patients who develop inhibitors to either the plasma-based or recombinant factor treatments are difficult to treat. Likewise, acquired hemophilia is an autoimmune disorder where the body naturally attacks clotting factors, usually factor VIII. Treatment for this disease is similar to that for hemophilia patients with inhibitors (Holme et al. 2005). Since the body mounts an immune response against the injected clotting or naturally present factors and destroys them, hematologists must use treatment methods to circumvent this occurrence.
ACTIVATED PROTHROMBIN COMPLEX CONCENTRATES
EFECTIVENESS
Activated Prothrombin Complex Concentrates (aPCC) contain Factor Eight Inhibitor Bypassing Activity (FEIBA) (Mannucci 2003) and have been used to treat hemophilia patients with inhibitors for the past 30 years (Luu and Ewenstein 2004). FEIBA works by activating the synthesis of thrombin by stimulating prothrombinase (Turecek et al. 2004), thereby bypassing the synthesis of factors IX and VIII (Sjamsoedin et al. 1981). FEIBA contains precursor hormones to clotting factors FVII, FIX, FX, and prothrombin but only contains trace amounts of the factors themselves. Therefore, no immune response is seen with this product. Although FEIBA is synthesized from human plasma, it undergoes a stringent, vapor-heated purification process and Turecek et al (2004) suggest that there are no reported cases of viral transmission from this product.
Negrier and coworkers (1997) reported on a study of FEIBA use in 60 hemophiliacs with inhibitors. The product was used in 433 bleeding events. The study found that FEIBA was successful at stopping bleeds in 81% of patients. Similarly, Sjamsoedin et al. (1981) report on a 64% success rate for the 15 patients in their trial which lasted 15 months. Although these results are promising, this rate is still lower than the effectiveness rate of multiple recombinant factor infusions (Mannucci 2003). Further, Hayashi et al. (2004) noticed that some patients experienced resistance to treatment over a prolonged period of time.
ASSOCIATED PROBLEMS
One of the main problems associated with the use of FEIBA is the risk of cardiovascular complications. Luu and Ewenstein (2004) noted that every 4 in 100,000 FEIBA injections led to severe cardiovascular disturbances caused by thrombi (blood clots). However, they claim that not only did most of patients with this problem overdose on the medication, but also they had genetic predispositions to cardiovascular disease. Mizon et al (1992) also report on a case where a patient experienced a myocardial infraction after 2 infusions with FEIBA. Their study further highlights the possible hazards of this treatment, especially if risk factors for cardiovascular disease are already present. Further, Sjamsoedin and colleagues (1981) report on a temporary loss of liver function after FEIBA infusions in 9 of their 15 patients. Additionally, since there are currently no available monitoring devices for FEIBA in vivo, it is difficult to measure patient response to the drug (Young 2006).
Although there have been few reported incidences of viral transmission with FEIBA (Turecek et al. 2004), a theoretical risk of infection is still possible since it is made from human tissue.
AVAILABILITY AND ACCESSIBILITY
Although the unit price for this type of treatment as compared to the treatment of patients without inhibitors is relatively similar (Chang et al. 1999), the need for repeated infusions with FEIBA due to its lower success rate increases the cost of treatment significantly. In 68% of patients with inhibitors, another method of treatment is more cost-effective (Joshi et al. 2006).
RECOMBINANT FACTOR VIIa
EFFECTIVENESS
Activated recombinant factors VII (rFVIIa), or Novoseven, function by binding directly or in conjunction with other clotting factors to platelets at the site of trauma (Monroe et al. 1997). rFVIIa may activate thrombin and other factors downstream of FVIII and FIX, thereby bypassing the immunoactivating step in coagulation. In a trial (Shaffer and Phillips 1997) of 67 bleeds, an 85% effectiveness rate has been reported with this drug in patients with inhibitors. A larger-scale study conducted by Key et al. (1998) reports on a 92% success rate in the 614 bleeding episodes that were treated with this method.
rFVIIa has been shown to effectively stop bleeding episodes with one dose as opposed to treatment with FEIBA (Joshi et al. 2006). Re-bleeds after infusion were associated with 50% of patients in the Key et al study. However, patients using rFVIIa have been shown to be resistant to treatment when the product is used over a prolonged period of time (Hayashi et al. 2004).
A Cochrane review on which treatment method arrests bleeding in people with hemophilia A with inhibitors has shown that there have not been appropriate trials to clarify the relative effectiveness of recombinant factor VIIa concentrate compared to human plasma-derived concentrates (Hind 2004).
ASSOCIATED PROBLEMS
The major complication associated with this treatment is myocardial infarction, which is reported by Sugg et al. (2006) and noted from other literature by Mannucci (2003). In the study by Sugg et al. (2006), 10% of patients using this treatment developed myocardial infarction. Other reported side effects included rash, vomiting (Mayer et al. 2005), fever (Mayer et al. 2005; Scharrer 1999), and blood clots (Key et al. 1998; Mayer et al. 2005; Scharrer 1999). However, side effects developed in fewer than 15% of patients in each case. No antibodies were found to be produced against rFVIIa (Key et al. 1998).
AVAILABILITY AND ACCESSIBILITY
Due to a reduced need for re-infusion of product, rFVIIa is overall, more cost effective than FEIBA (Joshi et al. 2006); however, each product's per unit price is comparable. Also, the most cost-saving dose of drug is recommended to 80 micrograms/kg as opposed to the 40 micrograms/kg and 160 micrograms/kg doses (Earnshaw et al. 2006).
IMMUNE TOLERANCE AND IMMUNOSUPPRESSIVE TREATMENTS
EFFECTIVENESS
Another method of combating inhibitors is to decrease the body's immune response to coagulating factor treatment. One approach is to make the immune system tolerant with either high-dose (Mannucci 2003) or low-dose (Mauser-Bunschoten et al. 1995) factor treatments. Mauser-Bunschoten et al. (1995) injected 25 U/kg of FVIII every other day for a period of 0.5 to 28 months into patients found to produce inhibitors against the factor. Patients were said to be immune tolerant when half the injected FVIII remained in the patients' blood while retaining its coagulating activity and the inhibitor concentration decreased below a set value. Twenty-one out of the 24 patients in this study reached immune tolerance through this method. High-dose factor treatment has been successful in 70% of patients (Manno 2005).
Drugs that suppress the immune system are also effective at decreasing the destructive response of the body towards circulating coagulating factor. Rituximab is an anti-CD20 antibody that destroys existing B-cells which are present in immune response (Wiestner et al. 2000). In a study (Stasi et al. 2004) of 10 patients with inhibitors to FVIII who received rituximab treatment, 8 patients were found to undergo remission. However, 3 patients later relapsed and researchers noted that the most successful treatment option was a combination of rituximab and other immunosuppressive agents.
Common immunosuppressive agents used to control inhibitors include corticosteroids, cyclophosphamide, immunoglobulins (Ig) (Berezne et al. 2006), and prednisone (Yee et al. 2000). These drugs can be used alone or in combination to reduce immune activity. Intravenous Ig in combination with prednisone has been shown to raise platelet levels for a period of 2 to 6 weeks with one infusion (Manno 2005). A combination of prednisone and cyclophosphamide has been found to be an effective treatment option as well (Yee et al. 2000). Shaffer and Phillips (1997) achieved remission of inhibitors in the treatment of 9 patients with this method when the drugs were given daily during an average 12 weeks of treatment. Corticosteroids and cyclophosphamide are also a suitable combination for effective inhibitor reduction (Holme et al. 2005).
ASSOCIATED PROBLEMS
There are a few notable side effects that dissuade patients from the use of immunosuppressive treatments. Prolonged use of corticosteroids is associated with insomnia, weight gain, hypertension, and hyperglycemia. Ig infusions are sometimes accompanied by chills, rigor, and aseptic meningitis (Manno 2005). The most common concern is relapse of antibody production and need for further infusions. Also, the process by which immune tolerance is reached is a rigorous process that is often too strenuous for children (Manno 2005). Rituximab has been associated with adverse reactions of the mucus membrane and dermatitis as well as severe immunosuppression, making patients more susceptible to pathogens, possibly leading to dangerous infections (Bengtson et al. 2003).
AVAILABILITY AND ACCESSIBILITY
The process by which immune tolerance is induced is extremely costly due to the need for routine infusions with coagulating factors. This cost could be up to 2.5 times the cost of regular treatment (Teitel 2006). Infusions with Ig are also expensive (Manno 2005). Due to side effects, all immunosuppressive drugs require adequate monitoring which is readily available in the United States and Europe but not in countries with inferior health care systems.
GENE THERAPY
EFFECTIVENESS
Treating hemophilia with gene therapy appears promising because the disease is caused by a single gene defect and because only a small increase (5%) (Gan et al. 2006) in gene product could essentially transform a severe form of hemophilia into a mild one. Over activation of the gene up to 150% of its action has also not caused any adverse effects (VandenDriessche et al. 2001). Another advantage is that although clotting factors are made in the liver, they can be synthesized in a wide variety of cells (High 2006). With continuous supply of gene product, gene therapy could potentially cure hemophilia (VandenDriessche et al. 2001). Gene therapy has been successful in greatly improving, if not curing, hemophilia in dogs (Chuah et al. 2001) and in mice with knock-out mutations for the coagulating factor genes.
There are generally two approaches to gene integration into cells. Genes can be integrated into highly reproducing stem cells so that all the daughter cells express the gene and its product. The other approach is to integrate genes into long-living cells such as skeletal muscle, cardiac muscle, and central nervous system cells that will be present in the body long enough to continuously express the target genes (High 2006). Cell types that have been considered for gene integration include fibroblasts, epithelial cells, endothelial cells, and bone marrow cells. Target cells may be removed from a patient, harvested in a culture medium, engineered to express a target gene, and then implanted back into the body (Mannucci 2003).
The procedures by which genes are delivered into the target cells are divided into two categories: viral-mediated and non-viral-mediated gene transfer. Generally, viral vector-mediated transfers have been found to be more efficient than non-viral-mediated transfers. Viral vectors are created by removing genetic material from viruses and replacing that material with the genetic material of the gene of interest. The machinery that incorporates genetic material into the host genome is already present in the virus. Viral vector transfers employed in coagulating factor integration have included the use of lentiviral (such as HIV), retroviral, and adenoviral vectors (VandenDriessche et al. 2001). The use of HIV-mediated transfer of FVIII has been successfully demonstrated in knock-out mice such that therapeutic levels of the clotting factor were produced (Kootstra et al. 2003). Adeno-associated viral vectors were used to transmit FIX into humans with a 10% restoration of clotting factor function in one hemophilia B patient (Ponder 2006).
Non-viral methods use naked DNA plasmids that not only include the gene of interest but also include genes encoding for protein products that will incorporate the target gene into the mammalian genome (Margaret et al. 2003). Yant et al. (2000) successfully transferred genes for FIX into mice through the non-viral method and achieved a greater than 5 month expression of clotting factor. Cells that were genetically engineered to express clotting factor genes can be administered to patients via grafting surgery or injections (VandenDriessche et al. 2001).
From 2001 to 2003, 5 clinical trials involving human hemophilia subjects and gene therapy were approved. In one of the trials, 13 patients with severe hemophilia A were given intravenous retroviral vector with the FVIII gene. However, the level of FVIII produced was only 1% of the desired level. In a second trial, the FVIII gene was administered to 6 patients through the non-viral method. Although levels of clotting factor rose in 4 out of 6 patients, levels of factor returned to pretreatment levels after a year of treatment. These two studies show the possible limitations of gene therapy including gene silencing and immunological response. Possible solutions to remedy these problems include the following: RNA repair, which will assist in the packaging of larger genes such as those for FVIII; genetically modified endothelial cells, which may prove to be better target cell for gene transfer; and genetically modified stem cells, which could potentially be stimulated to produce high levels of clotting factors (Margaret et al. 2003). As of yet, there have been limited human models that were able to produce continuous coagulating factor expression. Further, human models were unable to produce the same degree of coagulating factor production achieved in animal models (Mannucci 2003). Though gene therapy is promising, it is currently not a viable option for mass use among hemophilia patients.
ASSOCIATED PROBLEMS
Although animal models have shown some promising results, human models have not yet demonstrated the universal success of gene therapy (Pipe 2004). One of the biggest concerns with gene therapy, as with factor replacement therapies, is the production of inhibitors. The production of inhibitors in patients varies on the mode of transmission of the target gene, the target cells used in transmission, dose of vector used, and type of vector used (VandenDriessche et al. 2003).
Gan et al. (2006) suggest that introduction of gene therapy to newborn animals may prevent the formation of inhibitors; however this theory is more practical in animal models than in human patients. The immune reaction may be targeted against the protein product or the virus itself, most notably the capsid portion (Margaret et al. 2003). Since the body has probably encountered the types of viruses used in viral-mediated gene integration methods before, it has already formed antibodies against these viruses (VandenDriessche et al. 2001). The lack of continuous expression of gene product in most trials is most likely due to inhibitor formation.
Another associated problem relates to gene therapy as a whole. The risk of genetic mutation became realized when two children developed cancer 3 years after successful gene transfer. The possible culprit was the integration of the target gene into an oncogene, which codes for uncontrolled growth (High 2006). In a similar case, 2 infants who were treated with gene therapy at the age of 3 months both developed leukemia 2 years after therapy (Mannucci 2003). Possible solutions to mutagenesis of inserted genes include surrounding the gene sequence with "shielding" DNA, using an insertion vector that directs the target gene into specific region of the genome, and adding gene sequences that will excise a target gene from the genome if it becomes overactive (High 2006). Further, vector fragments have been found in the semen of patients treated with gene therapy, indicating that the vector may be passed on to progeny. More importantly, the rate of mutations is much higher in the germ line than in somatic cells, putting patients at higher risk for mutagenesis (Mannucci 2003).
Additionally, certain patients cannot be candidates for gene transfer procedures. Patients with liver disease cannot use genetically engineered hepatic cells (VandenDriessche et al. 2001). Further, if cells must be implanted into patients through surgical means, patients with severe hemophilia may not be able to withstand the surgical procedure.
AVAILABILITY AND ACCESSIBILITY
The use of gene therapy in the treatment of hemophilia may one day cure the disease; however, there are currently too many associated problems to make this treatment available to all patients (High 2006). Gene therapy is still undergoing clinical trials and is in very limited use by human subjects. Clinical trials are generally limited to patients with severe hemophilia who have had little success with other treatments. Additionally, therapies are very expensive and require strict supervision (Mannucci 2003).
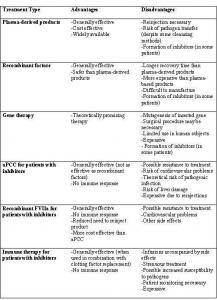
Table 1: Advantages and Disadvantages of Treatment methods of Hemophilia
DISCUSSION AND CONCLUSION
Each treatment described has many associated complications and side effects and thus, there is not a single treatment method that fits all hemophilia patients. Table 1 summarizes the advantages and disadvantages of each treatment method.
An important issue in management of hemophilia is whether to provide clotting factor concentrates regularly for the prevention of joint bleeds and the resulting deformities (prophylaxis) or to treat with the factors only when they bleed (on demand). However, a Cochrane review on the short and long term outcome of the prophylactic use of clotting factor concentrate in people with severe hemophilia A or B found that there is insufficient evidence from Randomized Controlled Trials (RCTs) to assess whether the prophylactic clotting factor concentrates decrease bleeding and bleeding related complications in hemophilia A or B, compared to placebo, or on-demand treatment (Sobart et al. 2006).
Gene therapy is currently unavailable for mass use (Pipe 2004), but may be a viable option in the future. This treatment method will most likely not gain popularity among physicians in the near future due to its limited testing in human models and extreme side effects including invasive DNA incorporation and cancer (Mannucci 2003). Although delivery of blood and blood products is the most commonly used treatment method in the world for hemophilia, its risks (Ludlam et al. 2006, Mannucci 2003) outweigh its generally high success rate and low cost. Plasma-derived product transfusion always carries some risk of infection transmission, even after cleansing (Ludlam et al. 2006), and since hemophilia patients require multiple transfusions through-out their lives (unlike a trauma patient, for example), their risk of contracting an infection statistically increases (Ludlam et al. 2006). Recombinant factors are generally favored by physicians in developed countries (Mannucci 2003) despite the fact that they are generally more expensive and still do not resolve the issue of inhibitor formation (Lusher et al. 1993). Based on the review of available evidence from the literature in this article, it is suggested that recombinant factors are the superior method of treating hemophilia patients who do not develop inhibitors. This treatment has proven to be extremely effective (Mannucci 2003, Lusher et al. 1993) and well tolerated in majority of patients (who do not develop inhibitors to treatment). After conducting a similar review, Mannucci (2003) came to a comparable conclusion deeming recombinant factors as the suggested mode of treatment for non-inhibitor producing hemophiliacs. However, it should be noted that Mannucci (2003) did not condemn blood-based products and assured patients who use these treatments that the risk of infection is currently only theoretical. Additionally, it should be suggested that further research be conducted on methods to make recombinant factors more affordable and accessible to patients outside developed countries. Patients with inhibitors are much more difficult to treat (Franchini et al. 2006) and management methods also have many adverse effects. Of the three treatment methods discussed for these patients, immune tolerance induction and immunosuppression are generally not favored due to their strenuous side effects (Manno 2005), cost (Teitel 2006), and need for close monitoring during treatment (Manno 2005). FEIBA and rFVIIa are comparable treatments in that they generally have a low risk of side effects (Luu and Ewenstein 2004, Sugg et al. 2006, Key et al. 1998) and similar price per unit of product. FEIBA success rate per infusion is less successful than rFVIIa and thus requires more infusions than rFVIIa and is therefore more expensive (Joshi et al. 2005). For this reason and the fact that FEIBA carries a slight theoretical risk of infection and is difficult to monitor (Young 2006), rFVIIa treatments are preferred for patients who develop inhibitors to routine recombinant factor treatments.
Despite these conclusions, choosing the right treatment for hemophilia is a matter that must be decided on a case by case basis. It is important to familiarize patients with the risks of hemophilia treatment, ultimately prompting them to demand safer methods of treatments (Ludlam et al. 2006). Both physicians and patients must understand the hazards of each treatment in order to make the best decision for the management of the disease.
REFERENCES
Barrow, T. et al. (2000) Reduction of the antigenicity of factor VIII toward complex inhibitory antibody plasmas using multiply-substituted hybrid human/porcine factor VIII molecules. Blood 95, 564568.
Batorova, A. and U. Martinowitz (2002) Continuous infusion of coagulation factors. Haemophilia 8, 170-177.
Bengtson, K. et al. (2003) Successful use of anti-CD20 (Rituximab) in severe, life-threatening childhood immune thrombocytopenic purpura. Journal of Pediatrics 143, 670673.
Berezne, A. et al. (2006) Rituximab alone or in association with corticosteroids in the treatment of acquired factor VIII inhibitors: report of two cases. Transfusion Medicine 16, 209-212.
Chang, H. (1999) The impact of inhibitors on the cost of clotting factor replacement therapy in Haemophilia A in Canada. Haemophilia 5, 247- 252.
Chuah, K. et al. (2001) Gene therapy for hemophilia. The Journal of Gene Medicine 3, 3-20.
Earnshaw, R. et al. (2006) Cost-effectiveness of recombinant activated factor VII in the treatment of intracerebral hemorrhage. Stroke 37, 2751-2758.
Ehrenforth, S. et al. (1992) Incidence of development of factor VIII and factor IX inhibitors in haemophiliacs. Lancet 339, 594-598.
Evatt, B. et al. (1998) Surveillance for Creutzfeldt-Jakob disease among persons with hemophilia. Transfusion 38, 817-820.
Farrugia, A. (2002) Evolving perspectives in product safety for haemophilia. Haemophilia 8, 236-243.
Franchini, M. et al. (2006) Inhibitors in mild/moderate haemophilia A: an update. Thrombosis and Haemostasis 96, 113-118.
Gan, U. et al. (2006) Genetic engineering for haemophilia A. Expert Opinion on Biological Therapy 6, 1023-1030.
Goudemand, J. et al. (2006) Risk of inhibitors in haemophilia and the type of factor replacement. Current Opinion in Hematology 13, 316-322.
Germann and Stanfield. Principles of Human Physiology. 2nd ed. (2005) Pearson Education. San Francisco. Pg. 510.
Green, D. (2006) The management of acquired haemophilia. Haemophilia 5, 32-36.
Hayashi, T. et al. (2004) Unresponsiveness to factor VIII inhibitor bypassing agents during haemostatic treatment for life-threatening massive bleeding in a patient with haemophilia A and a high responding inhibitor. Haemophilia 10, 397-400.
High, K. (2006) The leak stops here: platelets as delivery vehicles for coagulation factors. The Journal of Clinical Investigation 116, 1840-1842.
Hind, D. et al. (2004). Recombinant Factor VIIa concentrate versus plasma derived concentrates for the treatment of acute bleeding episodes in people with Haemophilia A and inhibitors. Cochrane Database of Systematic Reviews. Issue 2.
Holme, A. et al. (2005) Acquired haemophilia: management of bleeds and immune therapy to eradicate autoantibodies. Haemophilia 11, 510-515.
Hunter, N. et al. (2002) Transmission of prion diseases by blood transfusion. Journal of General Virology 83, 2897-2905.
Ingram, C. (1976) The history of haemophilia. Journal of Clinical Pathology 29,46979.
Joshi, V. et al. (2006) Pharmacoeconomic analysis of recombinant factor VIIa versus APCC in the treatment of minor-to-moderate bleeds in hemophilia patients with inhibitors. Current Medical Research and Opinion 22, 23-31.
Key, S. et al. (1998) Home treatment of mild to moderate bleeding episodes using recombinant factor VIIa (Novoseven) in haemophiliacs with inhibitors. Thrombosis and Haemostasis 80, 912-918.
Knobel, E. et al. (2002) Inhibitors in the Swedish population with severe haemophilia A and B: a 20-year survey. Acta Paediatrica 91, 910-914.
Kootstra, N. et al. (2003) Efficient production of human FVIII in hemophilic mice using lentiviral vectors. Molecular Therapy 7, 623-631.
Ludlam, A. et al. (2006) Clinical perspectives of emerging pathogens in bleeding disorders. Lancet 367, 252-261.
Lusher, M. et al. (1993) Recombinant factor VIII for the treatment of previously untreated patients with hemophilia A. Safety, efficacy and development of inhibitors. Kogenate Previously Untreated Patients Study Group. New England Journal of Medicine 328, 453459.
Luu, H. and B. Ewenstein (2004) FEIBA safety profile in multiple modes of clinical and home-therapy application. Haemophilia 2, 10-16.
Manno, S. (2005) Management of Bleeding Disorders in Children. Hemotology 416-422.
Mannucci, P. and E. Tuddenham (2001). The Hemophilias - From Royal Genes to Gene Therapy. N. English Journal of Medicine 344, 1773-1778.
Mannucci, P. (2003) Hemophilia: treatment options in the twenty-first century. Journal of Thrombosis and Haemostasis 1, 1349-1355.
Margaret, E. et al. (2003) Congenital Bleeding Disorders. Hemotology 559-574.
Mauser-Bunschoten, P. et al. (1995) Low dose immune tolerance induction in hemophilia A patients with inhibitors. Blood 86, 983-988.
Mayer, A. et al. (2005) Safety and feasibility of recombinant factor VIIa for acute intracerebral hemorrhage. Stroke 36, 74-79.
Meng, Y. et al. (2006) In vitro differentiation of mouse ES cells into hepatocytes with coagulation factors VIII and IX expression profiles. Science in China Series C: Life Sciences 49, 259-264.
Mizon, P. et al. (1992) Myocardial infarction after FEIBA therapy in a hemophilia-B patient with a factor IX inhibitor. Annals of Hematology 64, 309-11.
Monroe, M. et al. (1997) Platelet activity of high-dose factor VIIa is independent of tissue factor. British Journal of Haematology 99, 542-547.
National Heart, Blood, and Lung Institute. Hemophilia. (2006) Available at: www.nhlbi.nih.gov/.
Negrier, C. et al. (1997) Multicenter retrospective study on the utilization of FEIBA in France in patients with factor VIII and factor IX inhibitors. French FEIBA Study Group. Factor Eight Bypassing Activity. Thrombosis and Haemostasis 6, 1113-1119.
O'Mahony, B. and C. Black (2005) Expanding hemophilia care in developing countries. Seminars in Thrombosis and Hemostasis 31, 561-568.
Ozkan, O. et al. (2006) Microvascular free tissue transfer in patients with hematological disorders. Plastic and Reconstructive Surgery 118, 936-944.
Pipe, W. (2004) Coagulation factors with improved properties for hemophilia gene therapy. Seminars in Thrombosis and Hemostasis 30, 227-237
Ponder, P. (2006) Gene therapy for hemophilia. Current Opinion in Hematology 13, 301-307.
Reiter, P. et al. (2003) Texas lifestyle limits transmission of dengue virus. Emerging Infectious Diseases 9, 8689.
Saenko, L. and M. Ananyeva (2006) Receptor-mediated clearance of factor VIII: implications for pharmacokinetic studies in individuals with haemophilia. Haemophilia 4, 15-22.
Scharrer, I. (1999) Recombinant factor VIIa for patients with inhibitors to factor VIII or IX or factor VII deficiency. Haemophilia 5, 253-259.
Shaffer, G. and D. Phillips (1997) Successful treatment of acquired hemophilia with oral immunosuppressive therapy. Annals of Internal Medicine 127, 206-209.
Sjamsoedin, J. et al. (1981) The effect of activated prothrombin complex concentrate (FEIBA) on joint and muscle bleeding in patients with hemophilia A and antibodies to factor VIII: A double-blind clinical trial. New England Journal of Medicine 305, 717721.
Stasi, R. et al. (2004) Selective B-cell depletion with rituximab for the treatment of patients with acquired hemophilia. Blood 103, 4424-4428.
Stobart, K. et al. (2006). Clotting factor concentrates given to prevent bleeding and bleeding-related complications in people with hemophilia A or B. Cochrane Database of Systematic Reviews. Issue 2.
Sugg, M. et al. (2006) Myocardial injury in patients with intracerebral hemorrhage treated with recombinant factor VIIa. Neurology 67, 1053-1055.
Teitel, J. (2006) Inhibitor economics. Seminars in Hematology 43, 14-17.
Turecek, L. et al. (2004) FEIBA: mode of action. Haemophilia 2, 3-9.
VandenDriessche, T. et al. (2001) Viral Vector-Mediated Gene Therapy for Hemophilia. Current Gene Therapy 1, 301-305.
VandenDriessche, T. et al. (2003) Gene therapy for the hemophilias. Journal of Thrombosis and Haemostasis 1, 1550-1558.
White, G. et al. (1998) Clinical evaluation of recombinant factor IX. Seminars in Hematology 35, 3338.
Wiestner, A. et al. (2000) Rituximab in the treatment of acquired factor VIII inhibitors. Blood 9, 342-346.
Yant, S. et al. (2000) Somatic integration and long-term transgene expression in normal and haemophilic mice using a DNA transposon system. Nature Genetics 25, 35-41.
Yee, T. et al. (2000) A survey of patients with acquired haemophilia in a haemophilia centre over a 28-year period. Clinical and Laboratory Haematology 22, 275-278.
Young, G. (2006) New approaches in the management of inhibitor patients. Acta Haematologica 115, 172-179.