
Author: Charles E. Schiaffo
Institution: University of Connecticut
Date: October 2006
Abstract
We have recently defined a concise, three-step synthesis to carbohydrate-based oxepines from pyranose lactols, where the key step involved a Schrock catalyst mediated ring closing metathesis (RCM) reaction. A sub-family of diene substrates could be cyclized using the Grubbs second-generation catalyst, although the yields were low (25-30%) and a side product, tri-O-benzyl glucal was observed (~10%). The present study was undertaken to find a cleaner and more efficient route of synthesizing oxepines. The strategy was to focus on the suppression of the side reaction during RCM that leads to glycal side products; this was done in three ways. First, RCM using Grubbs second-generation catalyst in the presence of 1,4-benzoquinone was attempted. Second, purified (via column chromatography) Grubbs second-generation catalyst and 1,4-benzoquinone were used to perform the RCM. Finally, the use of purified Grubbs II alone was used to accomplish RCM. The goal was to test each of these methods on 4,5,7-tri-O-benzyl-1,2,3-trideoxy-6-O-vinyl-D-glucohept-1-ene. The synthesis of 5,7-benzylidine-4-O-benzyl-6-O-vinyl-glucohept-1-ene via a nine-step synthesis is also described.
1. Introduction
It is well known that pyranose carbohydrates are biologically active. Septanose carbohydrates are seven membered sugars that appear to be similar to naturally occurring six membered sugars. Therefore, it is believed that septanose carbohydrates should be biologically active. This study was an attempt to create a new and more efficient route to carbohydrate-based oxepines, such as 4,5,7-tri-O-benzyl-1,2,3-trideoxy-D-glucohept-1-ene. These molecules are donors in the synthesis of septanose carbohydrates.

article_816_order_1
The synthesis of carbohydrate based oxepines using the Schrock catalyst has been well documented. The Schrock catalyst has been very effective at performing Ring Closing Metathesis and when the reactions are run the conversion is effective and in good yield1. However, this catalyst is also atmospherically sensitive and must be handled in a glove box. This led to the search for a catalyst capable of performing RCM that is easier to handle. While Grubbs second generation catalyst can perform RCM in atmospheric conditions it is not without its problems.
The initial investigation found that Grubbs II is not as good at performing RCM in comparison to Schrock. When Grubbs II is used in RCM on certain substrates, the diene will undergo RCM to the desired oxepine but the corresponding glycal is also formed as a byproduct. This formation occurs when the hydride impurity causes isomerization before RCM takes place. The formation of the glycal leads to a mixture of products that is difficult to separate by column chromatography. A solution to this problem has been proposed to be the use of quinones, in particular 1,4-benzoquinone, in reducing the isomerization of the diene to the glycal2. A hydride impurity in Grubbs II has been noted and is believed to be the reason for the glycal formation3.
The present study was undertaken to determine if the use of 1,4-benzoquinone and Grubbs II catalyst in a variety of combinations could be used to form oxepines while suppressing glycal formation. These new conditions will be tested on 4,5,7-tri-O-benzyl-1,2,3-trideoxy-6-O-vinyl-D-glucohept-1-ene. The synthesis of 5,7-benzylidine-4-O-benzyl-6-O-vinyl-glucohept-1-ene will also be described as a substrate in future RCM reactions. The below figure depicts the substrates that are being used in RCM reactions. With A being the substrate that RCM was done on and B being the molecule whose synthesis is described.

article_816_order_2
2. Results and discussion
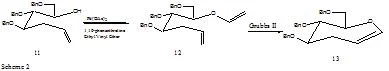
2.1 Ring closing Metathesis of 4,5,7-tri-O-benzyl-1,2,3-trideoxy-6-O-vinyl-D-glucohept-1-ene (14)
The synthesis of this molecule involved simple organic conversions.

article_816_order_3
The formation of 12 was done using previously reported methods5. The ring closing on 12 to 13 was done in four ways.

article_816_order_4
Table 1.
First, Grubbs II as provided by the manufacturer was used to measure a baseline of glycal formation. It was found that the composition of product was 3:1 oxepine to gylcal with diene still present. When the reaction was completed with Grubbs II from the bottle, 1,4-benzoquinone, the glycal in the product was practically nonexistent. There was, however, still a significant amount of diene present. Upon attempting to purify Grubbs II and 1,4-bezoquinone, glycal formation was still present along with some diene, though the glycal formation was reduced. Purified Grubbs II was used in order to test if the purification actually decreased glycal formation. The glycal formation was still present in a 1:1 mixture of glycal to oxepine. However, there was virtually no diene present in the product. At this time there is no explanation for these results.
2.2 Synthesis of 5,7-benzylidine-4-O-benzyl-6-O-vinyl-glucohept-1-ene (10)
The synthesis of 5,7-benzylidine-4-O-benzyl-6-O-vinyl-glucohept-1-ene was done using a nine-step synthetic plan.
article_816_order_5
The preparation of the molecule involved a straightforward synthetic chemistry through previously well-known reactions. The synthesis was initiated using commercially available α -D-glucose pentaacetate. In going from 1 to 2 no purification was done and the crude adduct was checked solely by 1H NMR. The crude adduct 2 was then carried directly through to 3, which was purified via column chromatography. The transformation from 1 to 3 accomplished an acetate elimination and shift. It was found that when 3 is azeotroped with dry toluene the reaction time is decreased from overnight to two hours. The product that formed from the azeotroped starting material did not experience decomposition. The product 5 was generated to put a thiophenyl in the 1 position as a protecting group. The conversion of 4 to 5 was efficient with no purification required and is completed as a precursor to the benzyladine reaction by replacing positions 3,4, and 5 with alcohols. Before 5 can be carried to 6, the remaining sodium methoxide needed to be neutralized or the conversion cannot occur. When, the crude adduct, 5 was filtered through Celite, it was found that a molar equivalent of p-toluenesulfonic acid (PTSA) needed to be added before the reaction would go. However, when crude 5 was filtered through silica gel it could be carried on to the next step with no extra PTSA needed. The transformation of 5 to 6 formed the benzyladine and occurred in high yield. When transforming 7 to 8, as a way to unprotect the 1 position, it was discovered that one equivalent NBS was more efficient than the published literature amount calling for six equivalents4. When six equivalent are used, 7 decomposes and neither the product nor starting material can be recovered. The conversion of 8 to 9 was done as a precursor to the vinyl ether formation by opening the chaing and forming an alkene. When completing the workup for 9 the excess phosphonium salt was removed by stirring the crude adduct in ethyl acetate and filtering. This process greatly reduces the impurities and enables 9 to be more easily purified by column chromatography. When converting 9 to 10 the current literature presented the best method5.
3. Conclusion
It has been found that the use of 1,4-benzoquinone is capable of effectively reducing glycal formation when using Grubbs II catalyst. Furthermore, the purification of Grubbs II is not necessary to reduce this formation. It has also been determined that the synthesis of 5,7-benzylidine-4-O-benzyl-6-O-vinyl-glucohept-1-ene can be completed using a nine-step synthesis.
4. Experimental Procedure
4.1 General Methods
Grubbs Second Generation Catalyst was bought from Aldrich and purified using a silica column and a 5:1 Hexanes to Ethyl Ether system. All of the NMR Spectra were run on either a Brüker 300 or 400 MHz NMR. Unless noted, reactions were performed at room temperature under an inert atmosphere of N2.
4.2. 1-Bromo-2,3,4,6-tetraacetate-D-Glucose (2)
α -D-Glucose pentaacetate was obtained from Acros Organics and used as is. In a round-bottom flask, HBr 30% in acetic acid (8.1mL, 153.6 mmol) was added to 1 (15g, 38.44 mmol). A homogenous yellow solution developed after one hour. The reaction stirred for 3hr and then 125 mL CHCl3 was added along with NaHCO3 and stirred for 1hr. Then another 15g NaHCO3 was added and stirred for 1hr. After the acid was quenched the yellow color of the mixture disappeared. When the mixture was filtered it yielded a clear solution that was rotary evaporated to dryness, yielding a white solid that was azeotroped three times with dry toluene before continuing on to the next reaction.
4.3. 1,3,4,6-tetraacetate-D-Glucose (3)
The azeotroped product (2) was dissolved in 300 mL of benzene in a round-bottom flask, to which AIBN(.662g, 64.04mmol) was added and stirred and refluxed for 30 min. Bu3SnH(12mL, 42.3mmol) was diluted to 30mL in benzene and added using a syringe pump at the rate of 2 mL/hr. The reaction was allowed to go 3 hr past the end of Bu3SnH addition. Purified using a silica gel column with 3:1 hexanes to ethyl acetate, resulting in a white solid 3 (60%).
4.4 3,4,6-triacetate-1-thiophenyl-D-Glucose (4)
1,3,4,6-tetraacetate-D-Glucose (6.8, 20.4mmol) was azeotroped 3x with dry toluene. It was then dissolved in 40 mL dry DCM in a round-bottom flask to which HSPh (2.4mL, 35.71mmol) was added. The reaction vessel was then placed under nitrogen and into an acetone/CO2 bath. After 10 min the BF3.OEt2 (8.78mL, 71.4mmol) was added drop wise. The reaction was allowed to go for 2.5 hrs at which time it was quenched with saturated NaHCO3. The reaction mixture was extracted with DCM and dried with Na2SO4, and the dichloromethane evaporated. A silica gel column used to purify with 3:1 hexanes to ethyl acetate as the eluent with an oil 4 (62%) resulting.
4.5 3,4,6-triol-1-thiophenyl-D-Glucose (5)
3,4,6-triacetate-1-thiophenyl-D-Glucose(4.661g, 12.2mmol) was added in a round-bottom flask to which 20mL sodium methoxide in methanol (2mg/mL) was added and stirred for 2 hrs. The solution was filtered through silica and washed with 800 mL of an 80% diethyl ether: 20% acetone solution. The solvent was evaporated and the product taken directly to the next reaction.
4.6 4,6-benzyladine-3-diol-1-thiophenyl-D-Glucose (6)
3,4,6-triol-1-thiophenyl-D-Glucose (2.789g, 10.88mmol) was dissolved in a round-bottom flask using DMF (60mL). Benzyladehyde dimethyl acetal (3.6mL, 23.9mmol) and PTSA (.21g, 1.088mmol) were added. The reaction was heated to 80ºC overnight. The reaction was quenched with 10 mL saturated NaHCO3, and evaporated to dryness. The mixtutre was redissolved in DCM, washed with saturated aqueous NaCl, and the DCM dried with Na2SO4. A silica column was run using 3:1 hexanes: ethyl acetate yielding a white solid 6 (58%).
4.7 4,6-benzyladine-3-O-benzyl-1-thiophenyl-D-Glucose (7)
4,6-benzyladine-3-diol-1-thiophenyl-D-Glucose (1.2g, 3.5mmol) was azeotroped 3x in dry toluene and dissolved in 20 mL DMF. NaH (.279g, 6.97mmol) and BnBr (.827mL, 5.22mmol) were added and the reaction was allowed to stir overnight. 10 mL H2O was added to the reaction mixture and evaporated to near dryness, redissolved in DCM, washed with saturated aqueous NaCl, dried with Na2SO4, and evaporated. A silica column was run using 3:1 hexanes: ethyl acetate, resulting in a white solid 7 (95%).
4.8 4,6-benzylidine-3-O-benzyl-1-diol-D-Glucose (8)
4,6-benzyladine-3-O-benzyl-1-thiophenyl-D-Glucose (1.3g, 3.0mmol) was cooled to zero degrees Celsius in a round-bottom flask. To the flask NBS (.54g, 3.03mmol) was added and stirred at 0ºC for 10 min and RT for 10 min. The reaction was extracted 3x with ethyl ether washed with H2O and Brine, dried with Na2SO4, and evaporated. Yielding a white solid 7 (60%).
4.9 5,7-benzyladine-4-O-Benzyl-6-diol-D-Glucohept-1-ene (9)
4,6-benzylidine-3-O-benzyl-1-diol-D-Glucose (.5g, 1.5mmol) was azeotroped 3x with dry toluene and dissolved in THF. The ylid was prepared by adding 1.6M BuLi (3.2mL) to triphenylphosphonium bromide (1.8g, 5.1mmol) at 0ºC and stirred for 20 min. The lactol was then cooled to 0ºC and1.6M BuLi (.92mL) added. The lactol was added to the ylid at 0ºC and stirred at RT overnight. The reaction was quenched with H2O and evaporated. Ethyl acetate was added to the crude adduct and stirred for 3 hrs, filtered, and evaporated. The adduct was put down a silica gel column using 3:1 hexanes to ethyl acetate, yielding 9 (67%).
4.10 5,7-benzylidine-4-O-benzyl-6-O-vinyl-glucohept-1-ene (10)
In a flame dried round-bottom flask ethyl vinyl ether (40mL) was added followed by 1,10-phenanthroline (.027g, .15mmol) and Pd(OAc)2 (.033g, .15mmol) were added and allowed to stir for 20 min. Then 5,7-benzyladine-4-O-Benzyl-6-diol-D-Glucohept-1-ene (.31g, .99mmol) in dry DCM (10mL) was added and heated to 40ºC for five days. After five days another one half of the original amount of all reagents except the benzyladine were added. The reaction was allowed to react for another four days. The reaction was evaporated and a silica gel column run using 3:1 hexanes to ethyl acetate. A white solid 11 (15%) was recovered.
4.11 4,5,7-tri-O-benzyl-1,2,3-trideoxy-6-O-vinyl-D-glucohept-1-ene (12)
In a flame dried round-bottom flask 40mL ethyl vinyl ether was added to which 1,10-phenanthroline (.057g, .032mmol) and Pd(OAc)2 (.071g, .032mmol) were added. This solution was stirred at RT for 20 min. After which 4,5,7-tri-O-Benyl-1,2,3-trideoxy-6-diol-D-glucohept-1-ene (.90g, 2.1mmol) in 10mL dry DCM was added. The reaction was allowed to reflux for 5 days. The reaction was evaporated and a silica column used to purify using 3:1 hexanes to ethyl acetate, yielding 12 (25%).
4.12.1 4,5,7-tri-O-benzyl-1,2,3-trideoxy-D-glucohept-1-ene (13)
In a round-bottom flask Grubbs II (.009g, .011mmol) from the bottle was dissolved in 3mL dry DCM. To this mixture 4,5,7-tri-O-benzyl-1,2,3-trideoxy-6-O-vinyl-D-glucohept-1-ene (.025g, .055mmol) was added and reacted for 24 hrs. The mixture was evaporated and a column run using 3:1 hexanes to ethyl acetate yielding 13 (16.5mg, 70%) a colorless oil.
4.12.2 4,5,7-tri-O-benzyl-1,2,3-trideoxy-D-glucohept-1-ene (13)
In a round-bottom flask Grubbs II (.009g, .011mmol) from the bottle and 1,4-benzoquinone (.012, .11mmol) were dissolved in dry DCM. To this mixture 4,5,7-tri-O-benzyl-1,2,3-trideoxy-6-O-vinyl-D-glucohept-1-ene (.03g, .06mmol) was added and reacted for 24 hrs after which more Grubbs II ( .004g, .0055mmol) from the bottle was added and allowed to react for another 24 hrs. The mixture was evaporated and a column run using 3:1 hexanes to ethyl acetate yielding a 13 (4.1mg, 17%). a colorless oil
4.12.3 4,5,7-tri-O-benzyl-1,2,3-trideoxy-D-glucohept-1-ene (13)
In a round-bottom flask purified Grubbs II (.009g, .011mmol) and 1,4-benzoquinone (.012g, .11mmol) was dissolved in dry DCM. To this mixture 4,5,7-tri-O-benzyl-1,2,3-trideoxy-6-O-vinyl-D-glucohept-1-ene (.025g, .055mmol) was added and reacted for 24 hrs after which more purified Grubbs II ( .004g, .0055mmol) was added and allowed to react for another 24 hrs. The mixture was evaporated and a column run using 3:1 hexanes to ethyl acetate yielding a 13 (8.2mg, 35%) a colorless oil.
4.12.4 4,5,7-tri-O-benzyl-1,2,3-trideoxy-D-glucohept-1-ene (13)
In a round-bottom flask purified Grubbs II (.009g, .011mmol) was dissolved in dry DCM. To this mixture 4,5,7-tri-O-benzyl-1,2,3-trideoxy-6-O-vinyl-D-glucohept-1-ene (.03g, .055mmol) was added and reacted for 24 hours after which more purified Grubbs II ( .004g, .0055mmol) was added and allowed to react for another 24 hrs. The mixture was evaporated and a column run using 3:1 hexanes to ethyl acetate yielding a colorless oil 13 (9.8mg, 42%).
Acknowledgements
The author would like thank Dr. Mark Peczuh for his invaluable help and guidance and Dr. Martha Morton (NMR) for her help in interpreting spectra as well as all the members of the Peczuh research group for their assistance. This work was supported by the National Science Foundation, the University of Connecticut, and the University of Connecticut Research Fund.
References
1. Peczuh, M. W.; Snyder, N. L. Tetrahedron Lett. 2003, 41, 4057-4061.
2. Hong, S.H, Sanders, D.P., Lee, C.W., Grubbs, R.H. Personal Communication.
3. Hong, S.H., Day, M.W., Grubbs, R.H. J. Am. Chem. Soc. 2004, 126, 7414-7415.
4. Oshitari, T., Shibasaki, M., Yoshizawa, T., Tomita, M., Takao, K.,Kobayashi, S., Tetrahedron, 1997,.53, 10993-11006.
5. Handerson, S., Schlaf, M. Org. Lett. 2002, 4, 407-409.