
Author: Joseph Jankolovits
Institution: University of Connecticut
Date: October 2006
Abstract
The bioconjugation of a protein with an N-hydroxysuccinimide ester is a protein modification technique used to create biomedical and biotechnological products. Some of the difficulties in this type of protein modification reaction are that proteins have many amines which react with N-hydroxysuccinimide esters, and the reaction solutions are difficult to analyze due to the complexity of the components. This study tests on a small scale whether controlling the pH could potentially be used to improve the site specificity of a protein modification reaction. In this experiment, the reaction rate between an N-hydroxysuccinimide ester and the N-terminal amine of a model peptide was measured by mass spectrometry to determine the reaction kinetics at various pHs. Mass spectrometry proved capable of fast and sensitive analysis of the reaction solution. The data showed that the reaction of a peptide with an N-hydroxysuccinimide ester is pH dependent. This is attributed to different degrees of the protonation of the amine. The pH dependence of the reaction between an N-hydroxysuccinimide ester and the N-terminal on a peptide could potentially be applied to site-specific protein modification.
Introduction
Protein modification is a valuable biochemical process in which a protein is altered to change its structure or function. Protein modification is used in many areas, including protein therapeutics, protein engineering, and protein labeling (Sheffield, 2001; Ojida et al., 2005; Cornish et al., 1996; Shibata et al., 2005; Lecolley et al., 2004). Typically, protein modification occurs on the reactive functional side chains on amino acid residues like those on lysine or cystine, or on the reactive terminal ends of the protein (Tamilarisu et al. 2001). N-hydroxysuccinimide (NHS) esters are commonly used in protein modification as they react readily with primary amines at room temperature or lower (Pierce, 1998).
Generally, protein modifications are meant to be site specific, meaning the modification is only meant to react at one particular reactive group. Site specificity is important for minimizing unwanted side reactions in protein modification and obtaining homogeneous products (Wang and Walfield, 2005). Protein modification techniques that enhance site specificity would make the production of biomedical and biotechnological products easier, faster, and less expensive. However, proteins are very large and have many reactive functional groups, which makes site specific reactions difficult.
In this experiment, the potential applicability of precise pH control as a technique for enhancing the site specificity of the protein modification on the N-terminal amine will be assessed. Amines are in equilibrium with their unreactive protonated form, and since each amine will have a slightly different pKa for a variety of reasons including proximity to electron withdrawing groups and hydrogen bonding characteristics, the kinetics of protein modification reactions on amines may be pH dependent. If an amine on a protein reacts faster at a certain pH than other amines, then precise pH control could enhance the site specificity of the reaction.
Mass spectrometry has often been used in protein sequencing and kinetic analysis due to its sensitivity and ability to analyze and quantify multiple large compounds in one solution (Attwood and Geeves, 2004; Kolakowski and Konermanm, 2001; Norris et al., 2001; Steen and Mann, 2004; Bakhtiar and Tse, 2000). Therefore, mass spectrometry should be capable of measuring the reaction kinetics of this experiment.
To better manage analysis of the reaction rate, a model peptide with only one amine, on its N-terminal, will react with an NHS ester. Any pH dependence in the rate of the reaction between the NHS ester and model peptide will show the potential for using pH control as a method of site specific protein modification.
Reaction Scheme
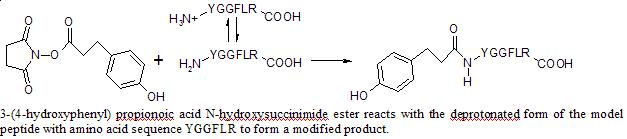
article_817_order_3
Experimental
A 10 mM solution of 3-(4-hydroxyphenyl) propionoic acid N-hydroxysuccinimide ester was prepared in anhydrous DMSO. 125 µL of this solution was added to 100 µL 0.1 mM of YGGFLR peptide in 100 mM phosphate buffer solution. The phosphate buffer solutions were prepared by combining, in proper stoichiometric amounts, 0.5 M solutions of monobasic and dibasic sodium phosphate. The 0.5 M solutions were prepared from sodium phosphate monobasic and dibasic powder. The reaction was quenched by putting 18 µL of the reaction solution in 125 µL of 1 M Tris HCl buffer at different times during a 60 minute period. 8 µL of 0.1 mM YGGFLK peptide was added as an internal standard. The solution was desalted in a C18 Micro spin column. The column was equilibrated with 2X 100 µL portions of MeOH followed by 2X 100 µL portions of 1% acetonitrile, 0.2 % formic acid in HPLC H2O (solvent A). The quenched reaction solution was run through the column and washed with 2X 100 µL portions of solvent A. The remainder was removed with 3X 100 µL portions of a 1:1 mixture of ACN and solvent A. The solution completely dried in a speedvac concentrator, and the remainder was dissolved in 160 µL of solvent A. The solution was injected by HPLC into the Q-Tof spectrometer, which uses an electrospray ionization source. Areas of the peptide and internal standard peaks from the ion chromatogram were measured and compared, and spectra were analyzed to ensure appearance of the expected product peak.
Results
The ratio of the peptide peak area to the internal standard peak area at t = 0 min was set as 100% peptide concentration. The ratio of the peptide peak area to the internal standard peak area at each time was normalized to the ratio at t = 0, and plotted to find the rate constant according to the equation:
k = - m (eq. 1)
where m is the slope of the plot of the natural logarithm of the concentration of peptide vs. time. k is taken from rate equation for the reaction (eq. 2).:
Rate = {k-1k2[H+][NHS ester]/(k1 + k-1[H+])}[Peptide] (eq. 2)
k1 and k-1 are the rate constants for the equilibrium of the peptide with its protonated form, and k2 is the rate constant for the reaction of the NHS ester with the deprotonated peptide. Due to the excess of NHS ester used, the equation was simplified to the pseudo-first order equation:
Rate = k[Peptide] (eq. 3)

article_817_order_1
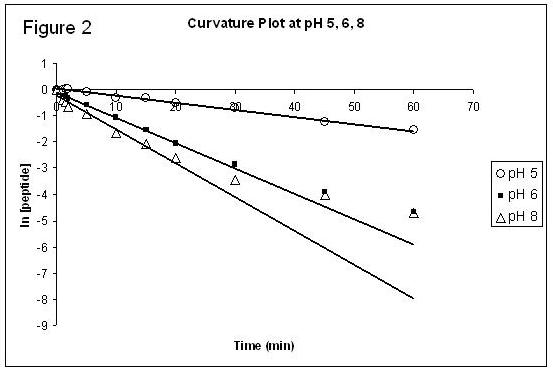
The experiment was performed at least twice at pHs of 5, 6, 7, and 8. Plots of the natural logarithm of the average concentration of the peptide vs. time at pH 5, 6, and 8 for the first 15 minutes of the reaction are shown below in Figure 1. The average measured k value at each pH is shown on the figure. Figure 2 shows the plots for the average concentration of peptide vs. time at pH 5, 6, and 8 over 60 minutes. The trendline on the figure is based on the first 15 minutes of the reaction to display the curvature of the linear plot at later times.
Discussion
The difference in the rate at each pH is most likely due to the varying degrees of protonation of the amine at each pH. At low pHs, the peptide's N-terminal amine is more protonated than at a higher pH. Since the peptide's deprotonated N-terminal amine is the limiting reagent, the reaction proceeds slower at lower pHs.
article_817_order_2
The rate constants were measured based on only the first fifteen minutes of the reaction because there was a loss of linearity at later times (Figure 2). The loss of linearity was caused by the competitive side reaction, the hydrolysis of the NHS ester. The curvature was the most dramatic at higher pHs, being very significant at pH 8 and not noticeable at pH 5. This is because the hydrolysis was much more prevalent in more basic conditions. The hydrolysis was significant enough to cause the loss of pseudo first-order rate properties at times after 15 minutes. The prevalence of the hydrolysis side reaction indicates that protein modification with NHS esters should not be run at exceptionally high pHs because the side reaction would effectively prevent the protein modification from occurring.
The most likely sources of error in the work came from assuming that the quality of the materials remained constant throughout the research. The NHS ester hydrolyzes rapidly with water, so even though steps were taken to minimize its contact with water vapor in the air, the stock of NHS could have still hydrolyzed over time. Also, the anhydrous DMSO could have absorbed water from the air, or the C18 micro spin column could have degraded in quality over time. It is for these reasons that the pH 7 data is not presented. The experiments at pH 7 were performed both at the beginning and end of the research. The rates from those experiments did not correlate well, and there was not enough time to repeat the experiment. Nevertheless, there were no indications of systematic error in the experiment, and any errors coming from the degradation of the materials would not be significant enough to warrant doubt on the broad conclusions drawn from the data.
Mass spectrometry was sensitive enough to analyze the microscale solutions, capable of distinguishing and quantifying each compound in solution and could easily handle the large mass compounds. Mass spectrometry was a very useful tool for measuring the kinetics of this peptide modification reaction, and should be used as the analytical tool in future experiments.
The results of the experiment indicate that there is a pH dependence in the rate constant of the peptide modification reaction of an NHS ester with an amine (Figure 1). The pH dependence of the reaction indicates that there is the potential for using pH control to enhance site specificity by setting the pH where the reaction at the desired site proceeds the fastest.
For future work, the experiment could be performed on more complicated systems. Since the reaction between the NHS ester and a peptide with one amine was pH dependent, the experiment should be run on a peptide with two amines to test for differences in the reaction rates on each amine. If there is a difference in the rates, the pH should be set where the reaction proceeds fastest on one amine to encourage a site specific reaction. From there, the experiment could be scaled up again to even more complicated peptides until the technique could be used on proteins as a method for improving the site specificity of protein modifications.
Acknowledgements
I would like to thank Dr. Xudong Yao for his guidance on this project; Alex Ramos, Yu Shi, and Hua Yang for their help in lab; The University of Connecticut REU program for giving me this opportunity; and the National Science Foundation for running the REU program.
References
[1] Sheffield, W.P. (2001) Modification of clearance of therapeutic and potentially therapeutic proteins. Curr. Drug Targets Cardiovasc. Haematol. Disord. 1(1):1-22.
[2] Ojida, A. et al. (2005) Suzuki coupling for protein modification. Tetrahedron Letters 46: 3301-3305.
[3] Cornish, V. et al. (1996) Site-Specific Protein Modification Using a Ketone Handle. J. Am. Chem. Soc. 118: 8150-8151
[4] Shibata, H. et al. (2005) Optimization of Protein Therapies by Polymer-Conjugation as an Effective DDS. Molecules 10: 162-180.
[5] Lecolley, F et al. (2004) A new approach to bioconjugates for proteins and peptides ("pegylation") utilising living radical polymerization. Chem. Commu. 2026-2027.
[6] Tamilarasu, N. et al. (2001) A New Strategy for Site-Specific Protein Modification: Analysis of a Tat Peptide-TAR RNA Interaction. Bioconjug. Chem. 12 (2): 135-138.
[7] Pierce Chemical Company. (1998) Instructions: NHS-Esters-Maleimide Crosslinkers. http://researchlink.labvelocity.com/protocols/protocol.jhtml;$sessionid$PXDOEFQAAAAN0QBICNWBNWQ?sourceId=32&path=0%7C30%7C38%7C940&nodeId=940&id=2377 (Accessed 6/17/05).
[8] Wang, C. and A. Walfield (2005) Site-specific peptide vaccines for immunotherapy and immunization against chronic diseases, cancer, infectious diseases, and for veterinary applications. Vaccine 23: 2049-2056.
[9] Attwood, P. and M. Geeves (2004) Kinetics of an enzyme-catalyzed reaction measured by electrospray ionization mass spectrometry using a simple rapid mixing attachment. Analytical Biochemistry 334: 382-389.
[10] Kolakowski, B. and L. Konermanm (2001) From Small-Molecule Reactions to Protein Folding: Studying Biochemical Kinetics by Stopped-Flow Electrospray Mass Spectrometry. Analytical Biochemistry 292: 107-114.
[11] Norris, A. et al. (2001) Kinetic Characterization of Enzyme Inhibitors Using Electrospray-Ionization Mass Spectrometry Coupled with Multiple Reaction Monitoring. Anal. Chem. 73: 6024-6029.
[12] Steen, H and M. Mann (2004) The abc's (and xyz's) of peptide sequencing. Nat. Rev. Mol. Cell. Biol. 5: 699-711.
[13] Bakhtiar, R. and F. Tse (2000) Biological mass spectrometry: a primer. Mutagenesis 15: 415-430.