

Author: Joseph L. Wezenter
Institution: Montclair University
Date: September 2005
Abstract
Given the current projections, maize will become the largest crop worldwide within a few years. It is vital that a crop this important have a well-defined genome. One hundred and thirty Bacterial Artificial Chromosome (BAC) maize sequences have been analyzed through various bioinformatics software, such as BLAST (Basic Local Alignment Searching Tool), GenScan, and Clustalx. BLAST analysis revealed that the maize genome is composed of diverse sequences that represent a variety of families of retrotransposons. These are segments of DNA that move from one location on a chromosome to another, resulting in genetic variation. Some mutations are phenotypically visible because of the color changes they represent within the endosperm. Many of these families appear to be closely related, and the most common retrotransposons are Ji, Huck, and Opie. Transposons were also present. The difference between transposons and retrotransposons lies in their replication mechanism. Retrotransposons use reverse transcription much like a retrovirus. When a physical map is created, these elements appear to be randomly distributed. However, due to the sequences' relationship it is not always possible to definitively categorize them, and only a small percentage of the sequences are predicted to encode proteins. Having a well-defined and fully-mapped genome provides a plethora of possibilities, including future gene manipulation to create a more robust version of maize.
Introduction
The goals for this project were to determine what Long Terminal Repeats (LTR) retrotransposons and what genes are present, to find the start and stop locations of both, and to ultimately create physical maps depicting their locations. This information provides an opportunity for gene manipulation, which may result in a more full-bodied plant.
LTR retrotransposons are responsible for a great deal of genetic variation. Copies of these segments are made much like that of a retrovirus. RNA copies are transcribed back into DNA using reverse transcription and then inserted back into the genome. This reinsertion may slightly alter a gene function, change the gene entirely, or have no effect.
In the 1940s Barbara McClintock discovered the first transposable element by observing certain markers in maize kernels, and their movement from one chromosomal location to another. By definition, transposons, or "jumping genes", are segments of DNA that can move around to different positions in the genome of an organism. McClintock found that the transposons were responsible for a variety of mutations (Jones 2005). These mobile elements were phenotypically visible because of the color changes they presented in the maize endosperm.
Currently, there are projects underway to increase kernel size and pathogen resistance, thereby reducing the amount of pesticides and herbicides needed. These desirable traits would augment crop yield from its already astounding annual harvest of 705 million metric tonnes worldwide, and reduce cost.
Maize is a staple in the diet of many, but it is consumed in several forms besides as a whole kernel. Maize oil is extracted from the kernel and maize is also a raw material in the manufacturing of starch. A complex refining process converts the majority of this starch into sweeteners, syrups, and fermentation products, including ethanol. Refined maize products, such as sweeteners, starch, and oil, are abundant in processed foods such as breakfast cereals, dairy goods, and chewing gum.

Table 1. 130 Zea mays BAC sequence. This table gives a breakdown of how many genes and LTRs were found in a given accession number. The summary table demonstrates the entire length of the one hundred and thirty sequence (18,893,776 bps). From this there were only 291 genes and 1088 LTRs found.
In the United States and Canada maize is typically used as animal feed, with roughly 80% of the crop fed to livestock (http://www.agbios.com/dbase.php?action=Submit&hstIDXCode=1&trCode=CYCL1). The entire maize plant, the kernels, and several refined products, such as glutens and steep liquor, are used in animal feeds. Feed made from the whole maize plant makes up 10-12% of the annual corn acreage. Livestock that feed on maize include cattle, pigs, poultry, sheep, goats, fish, and companion animals.
It is estimated that approximately 50,000 80,000 genes are present within maize's ten chromosomes. These ten chromosomes contain about 2.5 billion base pairs, and the genome consists of the combined genes from several different ancestors. Each chromosomal region generally has a duplicate on another chromosomal arm. Thus, the nucleus of a maize cell may contain two or more versions of identical or very closely related genes in different locations. Rice is one of the ancestors. For comparative purposes it only has 20,000 40,000 genes, 12 chromosomes, and only contains 400 million base pairs.
Given the cosmic importance of maize and the size of the genome, researchers are employing bioinformatics to become more efficient, much like the crops they are trying to advance. Such technological advances allow for better time management and increased productivity.
Materials & Methods
Obtaining BAC Sequences

Table 2. LTR Frequency. This table gives a numerical breakdown of the most common LTRs found. Ji was the most common with 371 occurrences, followed by Huck and Opie with 267 and 212 respectively. The remaining 238 LTRs were assorted.
130 Zea mays BAC sequences were examined (Table 1). These are DNA fragments that are inserted into a bacterial cell and allowed to replicate. This provides a fast, effective way to produce copies of your sequences. The GenBank accession numbers provided were used as a query for the web-based database of NCBI (National Center of Biotechnology Information) (http://www.ncbi.nlm.nih.gov/). The category Nucleotide was selected and the accession number was entered. This provided the raw DNA sequence in the FASTA format, which was saved as a text file, and served as the basis for most of the analysis.
GenScan Analysis
With the raw sequence now saved, another web-based piece of software called GenScan was used. This runs from a server at MIT (http://genes.mit.edu/GENSCAN.html). GenScan predicts the locations and exon-intron structures of genes in genomic sequences from a variety of organisms such as maize, vertebrates, and Arabidopsis. By being organismspecific, the results are more precise. The FASTA sequences were loaded and maize was selected as the organism. From this the one letter amino acid sequences of the predicted genes were generated and uploaded into BLASTp (protein-protein) of the NCBI website for further analysis.
BLASTp Analysis
BLASTp is a rapid sequence comparison tool that uses a heuristic approach to construct alignments by optimizing a measure of local similarity (Tatusova et al 1999). As previously stated, the one letter amino acid sequence of the predicted genes was uploaded into BLASTp (http://www.ncbi.nlm.nih.gov/BLAST/) and Zea mays was selected as the organism of interest. However, as parameters were set up, only sequences containing 200+ amino acids were analyzed, and results were only accepted if the E-value was e-20 or less. The E-value represents the theoretical number of false hits per sequence query, with zero being the best possible outcome. The results from BLASTp include a graph with different alignment scores of genes and pseudogenes or retrotransposons. Pseudogenes are genes bearing close resemblance to known genes at different loci, but rendered non-functional by additions or deletions in the structure that prevent normal transcription or translation. Genes were the information of interest.
The Institute for Genomic Research (TIGR) Analysis
TIGR (http://www.tigr.org/tdb/e2k1/plant.repeats/) is another piece of web-based software. It is a collection of curate databases containing DNA and protein sequences, gene expression, cellular role, protein family, and taxonomy of microbes, plants, and humans (TIGR 2005).

Figure 1. Graph of LTR Frequency. Graphical depiction of the data presented in Table 2.
Within TIGR the Plant Repeat Database was used. In plants the ploidy levels and repetitive sequences contribute significantly to the genome. Therefore, a number of different repetitive sequences have been reported in the plant genome. These can be classified into super-classes, classes, and subclasses based on structure and sequence composition (TIGR 2005).
Some of the transposable elements include retrotransposon, transposon, and MITEs (miniature inverted-transposable elements). Therefore, TIGR was used to find all the retroelements that were present in the BAC end sequences of maize. The only drawback of TIGR is that it does not provide the start and end locations of the retroelements or the nucleotide sequences of the retroelements. Therefore, a BLASTn search was also performed in order to find and copy the nucleotide sequences of those retroelements.
BLASTn Analysis
BLASTn (nucleotide nucleotide) is a similar program to BLASTp; however, the algorithm finds similar sequences by generating an indexed table or dictionary of short sequences called "words" for both the query and the database (Tatusova et al 1999). This is very helpful for finding retroelements that were not shown by TIGR.
LTR_STRUC Analysis
LTR_STRUC is a program that identifies and automatically analyzes LTR retroelements in the genome databases by searching for structural features characteristic of such elements (McCarthy et al 2002). For LTR retrotransposon families with low sequence homology to known queries or families with atypical structure, the LTR_STRUC has significant advantages over conventional search methods (McCarthy et al 2002). The BAC sequences were run through the program, which generated valuable information. The information generated included a report file. These files provide an automated analysis of each transposon location: putative LTRs, 5', and 3'. The whole transposon sequence was checked in BLASTn in order to get their proper names.
BLAST2 Analysis

Figure 2. Copia and Gypsy element familes. Here is a tree diagram depicting element type. Ji, Opie, Prem, Victim, Fourf, and Hopscotch are all Copia elements. Huck, Grande, Cinful Tekay, Milt, Reina, and Magillan are all Gypsy elements.
The findings from the other software were run through BLAST2 for pair-wise comparison with relative BAC sequences in order to obtain the proper start/stop coordinates. BLAST2, a new BLAST based tool, can be used efficiently to compare two proteins or nucleotides. (Tatusova et al 1999) This is very useful because it allows for the comparison of only two sequences that are already known to be homologous. . In such cases, searching the entire database would be unnecessarily time-consuming.
ClustalX Analysis
ClustalX is a multiple sequence alignment program for nucleotide and protein sequences. ClustalX uses a method called pair-wise progressive sequence alignment. This heuristic method first does pair-wise sequences alignment for all the sequence pairs that can be constructed from the sequence set. A dendrogram (guide tree) of the sequences is then done according to the pairwise similarity of the sequences. Finally a multiple sequence alignment is constructed by aligning sequences in order, defined by the guide tree. Therefore, Clustalx was used as the last step, when a complete alignment was performed. Once the alignment was completed, the program generated a NJ-tree (neighbor joining tree) in which it grouped together the LTRs that were similar in sequence.
Results
The focus of this project was LTRs. From the 18,893,776 bps analyzed there were 1088 LTRs and 291 genes. From this the LTR density and gene density were calculated. The values were 17366 bp per LTR retrotransposon and 64927 bp per gene respectively. This means there are more LTRs in a given length of base pairs when compared to genes. From this it was determined that LTRs make up approximately 95% of the total length analyzed, with genes making up the final 5%. This is consistent with Meyers et al 2001 findings. The function of the genes varied more and most of the sequences did not encode for any enzymes. (Table 1).
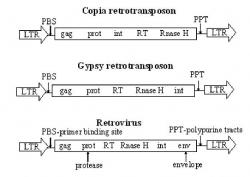
Figure 3. Structural comparison of Copia, Gypsy, and Retroviruses. This figure demonstrates the structural differences between Copia and Gypsy element, and then compares them to retroviruses. Gag is an intracellular packaging of the RNA transcript. EN is an endonuclease associtated with the integration into the genome. RT is a reverse transcriptase. Prot is a protease. Int is an intergase that mediates the specific DNA cleavage required for excision and integration. When these elements are compared to retrovirus it is apparent that they are very similar. Retroviruses contain all these components and one more; Env which is an envelope protein.
The most common LTRs found were Ji, Huck, and Opie. Combined, these three made up 78% of the 1088 total LTRs. Ji was the most common with 371 occurrences, followed by Huck and Opie with 267 and 212 respectively. The remaining 238 LTRs were assorted. (Table 2 & Figure 1).
LTRs are grouped in terms of their element type. The element can be either Copia-like or Gypsy-like. These structural differences are best explained pictorially (Figure 2 & 3). Copia and Gypsy retrotransposons contain the same structural components; only their order is different. All contain gag, EN, RT, Rnase, prot, and int. Gag is an intracellular packaging of the RNA transcript. EN is an endonuclease associated with integration into the genome. RT is a reverse transcriptase. Prot is a protease. Int is an intergase that mediates the specific DNA cleavage required for excision and integration. When these elements are compared to retroviruses, it is apparent that they are very similar. Retroviruses contain all the components of LTRs with one addition: Env, which is an envelope protein (Figure 3).

Figure 4. Phylogenic analysis of LTRs from AC148165 to AC148243. This diagram is comprised of the LTRs from accession numbers AC148165 to AC148243. LTRs of a common ancestor cluster together. A number is assigned after the element to identify the many variations (e.g. Ji1, Ji2, etc.). All the Ji's are grouped together. However, there is some deviation. Grande4 is in the middle of a Huck cluster.
A phylogenetic analysis was also preformed on these LTRs. This infers evolutionary history, and it is usually depicted in a TreeView diagram. This represents a pedigree of inherited relationships among molecules, organisms, or both. The distances are related to the degree of divergence between sequences. (Figure 4). As stated earlier, there are many variations of the same element, hence a different number would follow the LTR name (e.g. Ji1, Ji2, etc.). LTRs of a common ancestor cluster together. However, some LTRs appear to be out of place. Grande4 is within a Huck cluster. This was most likely do to an analytical error. It could have very easily been mislabeled.
The final step of this project was to create physical maps for all 130 BAC sequences(Figure 5). This was done using the start and stop locations generated by the various software. LTRs were distributed randomly throughout all of the sequences. Multiple pieces of software were used in conjunction with one another, not only to verify the results from each, but also to check for additional information. BLAST and LTR_STRUC gave different results. However, both are correct.
Discussion
It is known that transponsable elements have played an important role in facilitating major evolutionary transitions such as a significant increase in genome complexity (Bowen et al 2002). This genome complexity can give rise to great variability among species (Bowen et al 2002). Here 130 maize BAC sequences were analyzed using a variety of bioinformatics software. The data revealed that much of the maize genome is composed of a diverse group of LTR retrotransposons. This is supported by previous findings (Meyers et al 2001). Understanding an organism's genome and mapping it is vital for gene manipulation.

Figure 5. Physical map. This map is for AC150631. The total length of this BAC sequence is 178354 bps. This shows that LTRs are distributed randomly and that not every sequence will contain genes. BLAST and LTR_STRUC contained different results from one another, however both are correct. The positive and negative annotation refers to the direction in which it is transcribed.
The task of analyzing genomes is made easier by the emerging software. Gene finder programs like GenScan find open reading frames (ORF). ORF are long sequences of DNA that have no stop codons. . Here, axons within a sequence are assembled and, using computational methods, a putative protein is returned, creating frames. BLAST finds regions of similarity within a sequence by comparing it against a known database. This comparison can be performed with nucleotides (BLASTn) or with proteins (BLASTp). LTR_STRUC is another useful program. It finds complete LTRs within BACs, but has both some advantages and some drawbacks. There must be a 5' and 3' flanking sequence. Solo or partial LTRs will not be detected. Hence additional tools are needed to complete the annotation of the sequence. The alignment tool ClustalX finds homology, in this case the homology of LTRs families. Results are shown as a tree that represents a pedigree. LTRs of the same type are aligned together.
The homology trees also demonstrate which LTRs underwent the most change/mutation. In the case of the maize, it can be used to determine how similar the genome is to other organisms such as rice, beans, etc. Maize and rice came from a common ancestor. Sixty million years of evolution have altered genes, gene order, and genome size. Maize has 10 chromosomes, 50,000 - 80,000 genes, and is 2.5 billion bps in size. Rice has 12 chromosomes, 20,000 40,000 genes, and only 400 million bps.
With this being said, transponsable elements can easily out-replicate the genomes in which they reside, and potentially bring disastrous consequences to the host. However, like any evolutionary mechanism, there are organisms that develop certain defense mechanisms against it. For example, chromatin remodeling and methylation are two global silencing mechanisms that are thought to have evolved to repress the activity of transposable elements (Bowen et al 2002).
The physical maps created for the analyzed sequences provide valuable information. LTR retrotransposons are responsible for a great deal of genetic variation because of their ability to move from one location within the genome to another. Knowing the start and stop location of LTRs opens a window for manipulation; manipulation that could bring about increased annual harvests and reduce the amount of pesticides and herbicides needed, ultimately generating a superior crop.
References
AGBIOS. (2005)
http://www.agbios.com/dbase.php?action=Submit&hstIDXCode=1&trCode=CYCL1
Bowen, N.J. and I.K. Jordan. (2002) Transposable Elements and the Evolution of Eukaryotic Complexity. Current Issues in Molecular Biology. 4:65-76.
Burge, C. (2005) The New GENSCAN Web Server at MIT. Massachusetts Institute of Technology. http://genes.mit.edu/GENSCAN.html] http://genes.mit.edu/GENSCAN.html]http://genes.mit.edu/GENSCAN.html.
Fu H. et al. (2001) Transposons in maize. Proceeding of the National Academey of Scicenes. USA 98 8903-8908.
Jones, R.N. (2005) McClintock' controlling elements: the full story. Cytogenetics and Genome Research. 109:90-103.
MacNeish, R.A (1992) The origins of agriculture and settled life : By Richard S. Pp. 433. University of Oklahoma Press. Hardback £59.95 ISBN 0 8061 2364
McCarthy E, & McDonald, J, (2003) LTR_Struc: a novel search and
identification program for LTR retrotransposons. Bioinformatics 19 362-367
Meyers B.C, Tingey S, & Morgante M. (2001) Abundance,Distribution,and
transcriptional Activity of repetitive elements in the Maize Genome. Geneome
Research 11 1660-1676.
NCBI. (2005) BLAST. http://www.ncbi.nlm.nih.gov/BLAST/
Ouyang, S. and C.R. Buell. (2004) The TIGR Plant Repeat Databases: a collective resource for the identification of repetitive sequences in plants. Nucleic Acids Research. 32: Database issue.
Tatusova, T.A. and T.L. Madden. (1999) BLAST 2 SEQUENCES, a new tool for comparing protein and nucleotide sequences. FEMS Microbiology Letters. 174:247-50.
TIGR. (2005) BLAST search for plant repetitive sequences. http://tigrblast.tigr.org/euk-blast/index.cgi?project=plant.repeats.
http://www.agbios.com/dbase.php?action=Submit&hstIDXCode=1&trCode=CYCL1