

Author: Pattanayak Vikram
Institution: Biochemistry and Biophysics
Date: September 2005
Over the past decade, genetically modified organisms (GMOs) have been in the news for their potential benefits and harms to society. Before human cloning became an issue, the emergence of transgenic plants -- plants with foreign genes introduced to provide enhanced traits -- was the hot topic in biotechnology. These engineered crops seek to provide better flavor, disease resistance, and increased nutritional value just like traditional breeding methods (Kishore 1993; Ye 2000). Biochemical and molecular biological procedures provide the technology to genetically engineer plants, as well as the methods used to detect them.
One common method of engineering plants uses recombinant DNA technology mediated by the action of a soil bacterium, Agrobacterium tumefaciens, to transfer DNA from bacteria to plants (Davey 1989). Once the transformation has occurred, polymerase chain reaction (PCR) provides the ability to detect the genetically altered plants, allowing scientists to assess the success of the transformations, and in a different context, study the spread of transgenic crop species in the wild (Gachet 1999).
Creating Genetically Modified Plants
Agrobacterium tumefaciens contains a plasmid, a small circular piece of DNA that has its own origin of replication and is replicated independently of other nuclear material, that is key to its use in genetically modifying plants. This plasmid, called a tumor-inducing (Ti) plasmid, interacts with compounds released by fractured plant cells. When a wounded plant is exposed to A. tumefaciens, it integrates a stretch of its DNA, called transferred DNA (T-DNA), to the plant's genome by a recently elucidated mechanism (Van Attikum 2001).
Normally, the bacterium transfers its own T-DNA, but if the T-DNA is removed and replaced with another gene, A. tumefaciens can be used to introduce that gene into the plant genome, thereby providing a vector for scientists to engineer beneficial genes into plants (Horsch 1985). Studies have shown that native T-DNA genes are not necessary for this process. Inserted genes get transferred to plants as long as two repeated border sequences of 25 base pairs flanking the genes are present in the vector (Schell 1987). The Ti plasmid itself can only hold a 25 kilobase (kb) gene fragment, so it can only be used for small genes. It can also be somewhat difficult to work with (Hamilton 1996).

article_484_order_0
However, other vectors can be used to perform the transformation. Binary vectors, like the pBIN20 vector (Figure 1), are plasmids that contain the 25 base pair border sequences, allowing the new genes within them to be integrated into plant genomes, as well as marker genes that are used later in the process to select for successful gene transfer. The vectors also contain origins of replication for A. tumefaciens and Escherichia coli (Walden 1990), a bacterium commonly used in research. Therefore, the plasmids can replicate themselves in either E. coli or A. tumefaciens, allowing scientists to work with E. coli and then transfer the vectors to A. tumefaciens through bacterial conjugation, the process by which two bacteria exchange genetic information in the form of plasmids, when the genes are ready to be inserted into the plant genome.
Larger genes can be introduced to plants using bacterial artificial chromosomes (BACs). BACs are synthesized gene vectors based on a plasmid from E. coli (Griffiths 2000). BACs can have inserts ranging from 50 to 350 kb, allowing for the transfer of large genes or many small genes at once. The BACs used to transform plant cells are binary vectors, termed binary-bacterial artificial chromosomes (BIBACs)(Hamilton 1996). Like binary vectors, BIBACs contain gene markers as well as the two flanking boundary regions. Two commonly used gene markers in BIBACs and other binary vectors are neomycinphosphotransferase II (NPTII), which confers resistance to the antibiotic kanamycin, and hygromycin phosphotransferase, which gives resistance to the antibiotic hygromycin. BIBACs have been shown to successfully transfer the T-DNA insert between boundary regions to plants (Hamilton 1996), making them useful options for A. tumefaciens-mediated gene insertion.
article_484_order_1
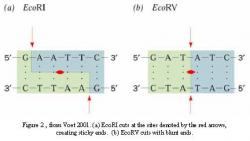
Transgenes can be inserted between the boundary regions of the BIBAC or other binary vectors through the use of restriction enzymes and recombinant DNA technology. Restriction enzymes cleave DNA at specific sites within a sequence of base pairs. The restriction enzyme EcoRI, for example, cuts both strands of double-stranded DNA between guanine (G) and adenine (A) whenever it sees the palindromic sequence GAATTC. It is called palindromic because one strand reads the same way as its complement when read from its 5' end to the 3' end. Because of this, the enzyme makes two cuts when it sees the recognition sequence, one on each strand (Figure 2a).
Another enzyme, EcoRV, cuts between thymine (T) and adenine every time it reads the sequence GATATC (Figure 2b). EcoRI and EcoRV represent two different types of restriction enzymes. EcoRI cuts with sticky ends, called "sticky" since each strand of cut DNA contains a four-base-pair overhang that can hydrogen bond to (combine with) a similar overhang on another molecule to form a double strand. EcoRV has no such overhangs and therefore cuts with what are known as blunt ends.

article_484_order_2
An enzyme that cuts with sticky ends can be used to insert a foreign gene into the vector of choice. If a restriction enzyme cuts the vector once, creating a gap between two sticky ends, and the same enzyme cuts on both sides of the coding region of the gene, it can be inserted into the vector. This is accomplished by cutting both with the enzyme and then "gluing" the gene fragment to the vector using a DNA ligase that helps hydrogen bond the complementary bases of the sticky ends (Figure 3). For example, this technique can insert a gene into the pBIN20 vector (Figure 1) if a site for the restriction enzyme Spe1, or any other sites found in the multi-cloning site (MCS), flanks it.

article_484_order_3
Once the appropriate vector is made and transferred to the A. tumefaciens bacterium, it must be integrated into the plant genome. Figure 4 shows how this transformation is carried out. First, disks are punched out from a plant leaf and incubated overnight with a culture of vector-containing A. tumefaciens in Luria broth. The disks are then placed in culture plates containing a medium that induces the leaf disks to start growing shoots, which are the precursors of the plant's stem and leaves. After a couple of days, these disks are transferred to plates, which contain antibiotics corresponding to the antibiotic-resistant marker genes placed in the vector. Only those plant cells that have integrated T-DNA from the vector will have antibiotic resistance. The medium therefore selects for the transformed plant cells by killing those that do not contain the vector. Once the transformed cells are selected, the growing shoots are transferred to a root-inducing medium, where they grow roots, and then to soil to grow into transgenic plants (Horsch 1985).
Detecting Genetically Modified Plants
Once the plants have grown in soil, they must be tested to determine if they contain a transferred gene. Although all of the grown plants are known to contain T-DNA from between the boundary regions of the vector through antibiotic selection, it is not yet known if the gene inserted into the vector during the recombinant step or not. The main method used to determine this is a fundamental procedure of biochemistry: polymerase chain reaction (PCR). PCR amplifies specific DNA sequences, creating millions of identical molecules using just one as a template.
PCR goes through many cycles of reactions, with each cycle containing three steps. First, the double-stranded DNA molecule separates into two strands when incubated at high temperatures (about 94° C), creating two complementary single-stranded DNA molecules. Next, the temperature is dropped to 55° C and two primers complementary to specific sequences within the DNA molecule bind. The primers are DNA oligonucleotides, short stretches of single-stranded DNA, which bind sequences that precede the 5' end of the region to be amplified on either strand. These primers bind to the complementary bases on the target DNA molecule, creating short double-stranded portions of DNA. Fianlly, the temperature is raised to 75° C, at which Taq polymerase, a heat-stable DNA polymerase isolated from Thermus aquaticus, catalyzes the extension of the primers on each strand. The polymerase recognizes double-stranded DNA and adds free nucleotides (dNTPs) to the 3' ends of the regions where the primers are bound (Voet 2001).
After the first cycle, one DNA molecule has become two molecules, since each separated strand has been bound by primer and extended by the polymerase. However, while these molecules contain the region of interest, the stretch between the two primers, they contain bases past the boundaries marked by the primers, since the polymerase does not know where the other primer is and extends the chain indefinitely. A second cycle of PCR amplifies the two newly synthesized strands to produce two more molecules. These molecules still have one strand that is longer than the target molecule. After the third cycle, there are eight molecules, two of which are identical to the target. Typically, PCR is carried out in 20 or more cycles, approximately doubling the amount of DNA each time, resulting in about 220 (approx. 1 million) copies of the target molecule (Gachet 1999).
PCR can be used to determine if a plant contains the desired transgene by extracting the plant's DNA and amplifying it. Since the sequence of the transgene is known and present only in transformed cells, PCR primers that recognize regions within the inserted T-DNA can be made (Gachet 1999). PCR is run on the extracted plant DNA with these primers, and a procedure called gel electrophoresis can determine its success.
In gel electrophoresis, DNA or protein samples are loaded onto a porous gel containing a network of agarose or polyacrylamide molecules. Electrodes are connected across the gel (often the gel is placed in a tank that has electrodes at its ends) causing a current to pass through it. DNA carries a negative charge, so it moves through the gel toward the positive electrode. The DNA encounters agarose or polyacrylamide molecules that hinder its path, with smaller molecules getting past these obstacles more quickly than larger ones. Since many molecules are placed onto the gel, they will separate according to size and form bands. The distances they travel are proportional to their molecular weights. The resulting separated bands can be detected using staining techniques or through the use of UV light that illuminates a fluorescent dye molecule bound to the samples before they are run.
The visualizing methods have sensitivities on the order of one nanogram (Voet 2001), so only bands containing a large number of DNA molecules can be seen. If the genes were successfully transferred to the plant, the plant DNA would contain primer recognition sites and be amplified by PCR, producing enough molecules to be seen on the gel. If the gene is not present, the primers would not bind, and only one molecule would be in the sample, producing no band when stained because of the sensitivity of the procedure. Appropriate controls -- performing PCR on a molecule that the primers should not recognize as well as one of known sequence that they should recognize -- must be used to show that known sequences were amplified and visualized only if they contained regions corresponding to the primers (Quist 2001).
Detection of transgenic DNA in wild plants is a little more complicated. Since this requires detecting DNA of unknown sequence, many different primer targets must be chosen to give the best chance of amplifying a transgene, if present. Three categories of sequences are often targeted: regulatory sequences from transforming vectors, genetic markers used to select the transformed cells during the engineering process, and sequences within the transgenes themselves (Gachet 1999). One such primer target is the P35S promoter sequence of cauliflower mosaic virus, as it is found in many commercial transgenic constructs (Quist 2001).
Applications for Genetically Modified Plants
The biochemical techniques used to create different types of transgenic plants are similar for each plant. However, the applications of these plants are quite diverse. The two most widely used genetic modifications introduce a gene for a toxin from the bacterium Bacillus thuringienisis (Bt). Bt toxin has been used to provide insect-resistance to crops like corn and soybean. Another application introduces genes that code for herbicide resistance, allowing farmers to spray crops with chemicals that kill weeds without harming the crops themselves (Ferber 1999). Both of these alterations seek to increase the crops' yield by making them more likely to survive. Other modifications seek to improve flavor or add nutrients to crops. The Flavr-Savr tomato had a modification that sought to improve taste by introducing traits allowing tomatoes to ripen longer on the vine through the slowdown of an enzyme that causes rotting. It was eventually taken off the market because it was too soft to be transported to supermarkets without being damaged (Moffat 1998). Another crop, golden rice, is engineered to be rich in beta-carotene, a vitamin A precursor, in order to provide this required nutrient to people who eat rice as their staple food. Creating golden rice involves synthesizing a whole biosynthetic pathway by cloning many genes into rice endosperm (Ye 2000).
While there have been some successes, some traits are harder to engineer than others. Despite the creation of insect-resistant plants, plants with disease-resistance have been harder to create. Conferring disease resistance involves introducing genes that specifically recognize an invading pathogen. These genes have been shown to be ineffective, as pathogens have quickly evolved mechanisms to evade them. There is hope, however, that in the future introducing genes that target molecules essential to the pathogen will be more effective (Stuiver 2001). Ongoing research in other areas continues, with the advent of GMOs that produce polymers and pharmaceuticals not far off (Gachet 1999).
Debate Over the Use of Genetically Modified Plants
With their diverse applications, geneticallly modified plants could greatly benefit society. However, it is important to note that some believe they could also harm society. The same antibiotic-resistance marker genes scientists use to select for genetically modified cells, if present in food products, could spread to pathogens in the body. These pathogens would then have antibiotic resistance, making them harder to kill with traditional medicines.
Another potential harm comes from engineering herbicide resistance genes into plants. If these genes spread to wild, competing plant populations, they could create so-called "superweeds," unable to be killed by herbicides. Introducing insect-resistance genes could also be dangerous, as some data indicates that they can kill non-harmful insects, causing a potential change in the distribution of insect species in a given ecosystem.
Genetically modified plants can potentially harm more than just crop yields and insect populations. We do not conclusively know if humans who carry allergies to certain types of plants, like peanut allergies, would be allergic to food products containing inserted genes from an allergen. If they were allergic, extensive labeling or other methods would be needed to ensure their safety. So far, experimental evidence to date in all these areas lends support to both those who believe genetically modified plants are harmful and those who believe they are beneficial. Ultimately more studies need to be conducted to weigh these and other risks against the potential benefits of genetically modified plants (Ferber 1999).
Summary
The use of biochemical techniques has allowed foreign genes to be introduced into plants. Fundamentally, this involves recombinant DNA techniques and integration of genes mediated by the bacterium Agrobacterium tumefaciens. Different vectors can be used to accomplish this transfer, but all of them must include a pair of repeating 25-base boundary sequences found in A. tumefaciens. The DNA between these sequences, T-DNA, is transferred into the plant genome, where it can later be expressed. Polymerase chain reaction and gel electrophoresis can determine the success of the gene transfer by using primers to mark genes or other known sequences within the T-DNA. PCR is also used as a detection technique to determine if a plant is transgenic, aiding the understanding of the natural spread of transgenic crops in the wild.
Suggested Reading
Alberts, B., et al. Molecular Biology of the Cell: Third Edition. New York: Garland, 1994
Davey, M.R., E.L. Rech, B.J. Mulligan. "Direct DNA transfer to plant cells." Plant Mol. Biol. 133 (1989): 273-285.
Ferber, D. "Risks and benefits: GM crops in the cross hairs." Science. 286(1999): 1662-1666.
Gachet, E., G.G. Martin, F. Vigneau et al. "Detection of genetically modified organisms (GMOs) by PCR: a brief review of methodologies available." Trends in Food Science and Technology. 9 (1999): 380-388.
Griffiths, A.J.F., J.H. Miller, D.T. Suzuki, et al. An Introduction to Genetic Analysis: Seventh Edition. New York: W.H. Freeman, 2000
Hamilton, C., A. Frary, C. Lewis et al. "Stable transfer of intact high molecular weight DNA into plant chromosomes." Proc. Natl. Acad. Sci. USA. 93(1996): 9975-9979.
Hennegan, K.P., K.J. Danna. "pBIN20: An improved binary vector for Agrobacterium-mediated transformation." Plant Mol. Biol. Reporter. 16(1998): 129-131.
Horsch, R.B., J.E. Fry, N. Hoffman et al. "A simple and general method for transferring genes into plants." Science. 227(1985): 1229-1231.
Kishore, G.M., C.R., Somerville. "Genetic engineering of commercially useful biosynthetic pathways in transgenic plants." Curr. Opin. Biotechnol. 4(1993): 152-158.
Moffat, A.S. "Toting up the early harvest of transgenic plants." Science. 282(1998): 2176-2178.
Quist, D., I.H. Chapela. "Transgenic DNA introgressed into traditional maize landraces in Oaxaca, Mexico." Nature. 414(2001): 541-543.
Schell, J. "Transgenic plants as tools to study the molecular organization of plant genes." Science. 237(1987): 1176-1183.
Stuiver, M.H., J.H.H.V. Custers. "Engineering disease resistance in plants." Nature. 411(2001): 865-868.
Van Attikum, H., P. Bundock, P. J. J. Hooykaas. "Non-homologous end-joining proteins are required for Agrobacterium T-DNA integration." EMBO. 20(2001): 6550-6558.
Voet, D., J.G. Voet, and C.W. Pratt. Fundamentals of Biochemistry: Upgrade Edition. New York: John Wiley and Sons, Inc., 2001
Walden, R. and J. Schell. "Techniques in plant molecular biology - progress and problems." Eur. J. Biochem. 192(1990): 563-576.
Ye, X., et. al. "Engineering the provitamin A (-carotene) biosynthetic pathway into (carotenoid-free) rice endosperm." Science. 287(2000): 303-305