

Author: Nadeera Dawlagala
Institution: Smith College
Date: September 2005
Abstract
Some researchers claim nucleosomes disappear during transcriptional activation, and others claim nucleosomes reposition themselves. For example, the PH05 gene found in Saccharomyces cerevisiae requires nucleosome repositioning prior to transcription. This research focuses on the importance of nucleosome remodeling in transcription. A procedure developed in a previously published study used a photochemical reagent to cross-link a cysteine on the nucleosome, followed by nucleosome mapping on a sequencing gel. This was set to be repeated. However in order to do this, nucleosomes needed to be constructed with two mutated histones. The histones to be used in this experiment were of Saccharomyces cerevisiae, and these histones do not naturally contain the amino acid cysteine in their sequences. In order to perform the photochemical method, histones yH2B and yH4 needed to be mutated to code for a cysteine. The purpose of this experiment was to mutate these two histones and find their optimal time for expression. The histones yH2B and yH4 were mutated by designing primers and using the Stratagene Quickchange® Site Directed Mutagenesis Kit, followed by purifying the histones, and then expressing them using a protocol devised by another laboratory (Luger Lab). As a result, the yH4 histone was mutated to code for a cysteine instead of a serine on the 48th base pair, and the yH2B was mutated to code for a cysteine instead of a glutamine on the 60th base pair. The optimal expression for yH2B was found to be two hours. Although the photochemical method of cross linking to study nucleosome remodeling still needs to be completed, the histone mutation and expression needed to perform this procedure were carried out successfully.
Introduction
Understanding the purpose of a nucleosome and its histone components was essential to the problem of mutating the yH4 and yH2B histones and expressing them. The nucleosome participates in DNA compaction into higher order structures such as chromosomes (Wongwisansri and Laybourn, 2004). The "fundamental unit" of chromatin is the nucleosome (White et al., 2001). The nucleosome structure is about 10 nm in diameter and is composed of 147 bp of DNA that winds around an octamer of histones, which are proteins. The octamer is composed of histones H2A, H2B, H3, and H4. H2A and H2B form dimers, and H3 and H4 form dimers. Each H3/H4 dimer organizes about 30bp of DNA. Two H3/H4 dimers form a tetramer and organize 60bp of DNA (Wongwisansri and Laybourn, 2004). The octamer consists of the H3/H4 tetramer and two H2A/H2B dimers (Wongwisansri and Laybourn, 2004). An additional class of histone, called the linker histone or H1, is on the outside of the octamer and fastens the whole DNA-octamer structure together (Heller et al., 2001). The positive charge of the histones allows them to be attracted to the negatively charged phosphate groups on the DNA (Heller et al., 2001). Histones have long tails (containing lysine and arginine) constituting a quarter of their size and protruding from the core octamer structure. These tails interact with the surrounding nucleosomal DNA and other nucleosomes (Chromatin Organization: Nucleosomes, 2004). Histone tails are on the N-termini, and their modifications affect chromatin compaction and transcription.

Figure 1. Nucleosome. Model of a nucleosome that includes the octamer of four different classes of histones and 147bp of DNA (Chromosome Organization 2004).
Further research needs to be conducted on the process of nucleosome remodeling, which is why the histones needed to be prepared. Specifically, histones of the unicellular yeast Saccharomyces cerevisiae were used, since the larger project focused on studying nucleosome remodeling on the PH05 promoter. The PH05 gene of the unicellular yeast, Saccharomyces cerevisiae, participates in maintaining sufficient phosphate levels within the cell. Previous studies have discovered that the transcription of this gene requires the reconfiguration of the nucleosomes (Terrell, 2002). However when the gene was activated, about four nucleosomes on the promoter were lost or reconfigured (Terrell, 2002). This reconfiguration is called chromatin remodeling or nucleosome remodeling, which is the repositioning and modification of the nucleosomes on the promoter region through an ATP dependent process (Bartholomew and Kassabov, 2004). Nucleosome remodeling affects transcriptional activation and repression, DNA replication, and recombination (Wongwisansri, 2003). When a nucleosome is remodeled, it allows the nucleosomal DNA to be easily accessible to transcription factors and polymerases. Some researchers argue that nucleosome remodeling complexes involve the movement or sliding of the nucleosomes. Remodeling is a prerequisite for the activation of transcription, as in the PHO5 gene. However it does not always lead to transcription of a gene (Terrell et al., 2002).
We looked for the best way of mutating these histones and tried to find the optimal time at which a strain of E. coli cells (BL21 DE3) would express them. This was important for the purpose of using these two mutated histones to form two separate nucleosomes. These nucleosomes are to be used for a future procedure. Primers were designed to mutate the sequence of yeast histones H4 and H2B. The Primer Design Guidelines in the Stratagene Kit instructions (2003) were used to design the appropriate primers. Plasmid vectors for all histones, except H3, were pET28 (Figure 3). H3's plasmid vector was pET3a. This future procedure, devised by Blaine Bartholomew and Stefan R. Kassabov (2004), would map nucleosomes to study nucleosome remodeling. Their protocol involves attaching a photoreactive aryl azide to cysteines engineered at specific points on the nucleosome. UV radiation activates aryl azide cross-linking with the nucleosomal DNA. Treatment of the cross-linked DNA creates a break in the nucleosomal DNA, which can be mapped by foot printing/sequencing gels. Bartholomew claims that the photochemical method of mapping nucleosomes is preferable because there is no need to provide special reaction conditions for the cleavage agents, the cross-linking reaction is efficient and takes only milliseconds to occur, and the procedure allows intermediates in nucleosome remodeling to be studied (Bartholomew and Kassabov, 2004). Many researchers have attempted to map nucleosomes after nucleosome remodeling using techniques such as adding digestive nucleases and hydroxyl radical foot printing. Bartholomew and Kassabov (2004) argue that these methods "are ineffective for mapping translational positions of remodeled nucleosomes." Yeast histones do not normally have cysteines in their sequence. This is why two histones were picked ( yH2B and yH4) and were mutated to have a cysteine so that for the future project, the photoreactive aryl azide could attach to the cysteine region of the nucleosome.

Figure 2a. DNA sequence for yH4 wild type. DNA sequence for the yH4 prior to mutation. Letters in bold represent amino acids that the DNA codes for. Yeast histone H4 will be mutated to have the amino acid C, cysteine, on the 47th base pair instead of serine (S). Figure includes amino acid methionine, as 1st amino acid, which is why figure presents mutation on 48th base pair. Methionine will be removed in this protocol.
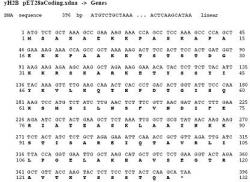
Figure 2b. DNA sequence for yH2B wild type. DNA sequence for the yH2B prior to mutation. Letters in bold represent amino acids that the DNA codes for. Yeast histone H2B will be mutated to have the amino acid C, cysteine, on the 59th base pair instead of glutamine (Q). Figure includes amino acid methionine, as the first amino acid, which is why the figure presents mutation on the 60th base pair. Methionine will be removed in this protocol.
The knowledge obtained from research on nucleosome remodeling contributes to our deeper understanding of molecular biology. Nucleosome remodeling plays a strong role in the transcription of some genes, which is why scientists notice its importance. The mechanism of nucleosome remodeling remains to be fully defined, and hopefully by studying this process we can understand how nucleosome remodeling regulates access to genes, particularly on the PHO5 promoter (Kassabov et al., 2003). Mutating and expressing the two histones is the first component in undertaking Bartholomew and Kassabov's mapping procedure.
Methods & Materials
Mutation
First, the creation of the yH4 mutant coding for a cysteine instead of serine on the 47th base pair was planned (Figure 2a). A cysteine was introduced into the 47th bp of yH4 using the wild type yH4 plasmid (vector pET28), the primers, and a Quick-change Stratagene Kit (1ul dNTP mix and 1ul of Pfu Turbo DNA polymerase) in PCR. Following PCR, 1 ul of Dpn I restriction enzyme was added, and the sample was incubated for one hour, which was followed by another incubation at 37°C for an additional hour. Following digestion of the original plasmid, 1 ul of the PCR products was added into 50 ul Xl-1 Blue Supercompetent cells. Following 30 minutes on ice and 45 seconds at 42°C, cells were grown in 0.5 mL SOC at 37°C for one hour with shaking at 225-250 rpm. 250 ul of cells were plated on LB plates that were also spread with 80 ul of 10 mg/ml kanomycin. Plates were then incubated at 37°C overnight. YH2B was mutated following the same protocol, but with primers that were designed to code for a cysteine instead of a glutamine on the 59th base pair (Figure 2b).
Plasmid purification
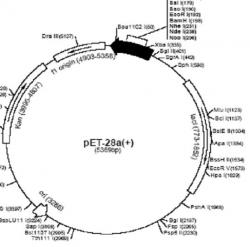
Figure 3. The pET-28a+ plasmid vector. The pET-28a+ plasmid vector was the vector for yH2B, yH2A, and yH4. During PCR (when amplifying and mutating the sequence for yH2B and yH4 plasmids), mutated primers were inserted at the restriction sites Xho1 158 and Xba1 335.
Colonies from incubated plates were picked and grown in 5 ml cultures in SOC broth and 20 ul of 10 mg/ml kanomycin. Colonies were grown overnight and then cultures were expanded to 5X5 ml cultures. Plasmids were then purified using the QIA Prep Spin Miniprep Protocol (2002), but with a few modifications: cells were first pelleted at 2000 rpm for seven minutes with a SS-34 Rotor and resuspended in 1250 ul Buffer P1. Additionally, after the elution of the DNA with 50 ul filtered water, DNA was microcentrifuged for two minutes at 8,000 rpm to remove excess buffer. Following purification, the plasmid DNA was run on a 1% agarose gel to determine the presence and concentration of DNA prior to sequencing. A DNA sample, -306 HTLV promoter DNA, served as the standard for comparison. The standard was diluted to 10ng/ul and 5, 10, 20, 50 and 100 ng of the standard was added to each well. Three different volumes of plasmid DNA (1ul, 2ul, 4ul) are loaded (Figures 4 and 5). After the concentration of the mutant plasmids was determined, they were sent to the facility (Macro molecular resources) to be sequenced (Figures 6 and 7).
Histone expression

Figure 4. 1% Agarose gel for yH4 mutant. After purifying the mutant plasmid, the plasmid was run on a gel with a standard to verify the presence of DNA and its concentration. Two yH4 plasmids were mutated in the experiment; A, B, and C represent one copy at different volumes, while A1, B1, C1 represent the other copy. The first copy was later the one chosen to be used to make the octamer. The standard (-306 HTLV promoter DNA) was added in different concentrations: 5, 10, 20, 50, and 100 ng. It was concluded that the yH4 plasmid DNA was about 100 ng/ul by comparing A' with the standard bands of DNA.
Mutant plasmids and previously purified wild type plasmids (provided by Laybourn lab) were transformed into the BL21 (DE3) Codon Plus strain of E. coli competent cells. This was done using the protocol in the Stratagene BL21 Codon Plus Competent Cell Instruction manual (2003), with a few modifications. The first modification was that 5 ul of the plasmid was diluted to 10 ng/ul and transformed into cells in 2X TY medium. In addition, cells were spread on LB plates that contained 34 ul of 34 mg/ml chloroamphenicol and 26.6 ul of 30 mg/ml of kanomycin. As a positive control, xH2B (a wild type Xenopus histone) plasmid obtained from the Luger lab was transformed into BL21 (DE3) pLysS cells (also contributed by the Luger lab) and spread on LB plates with 34 ul of 34 mg/ml chloroamphenicol and 16 ul of 50 mg/ml ampicillin. Plates were then incubated overnight at 37°C.

Figure 5. 1% Agarose gel for yH2B mutant. After purifying the mutant plasmid, the plasmid was run on a gel with a standard to verify the presence of DNA and its concentration. A, B, and C represent the different volumes of plasmid loaded (1 ul, 2 ul, and 4 ul). The standard (-306 HTLV promoter DNA) was added in different concentrations: 5, 10, 20, 50, and 100 ng. It was concluded that the yH2B mutant plasmid DNA was about 20 ng/ul by comparing A' with the standard bands of DNA.
The following unpublished protocol for histone expression was developed by the Luger lab. Six 20 ml culture tubes were prepared by adding the appropriate antibiotic (12 ul of 10 mg/ml kanomycin for yeast histones or 2.4 ul of 50 mg/ml ampicillin for the Xenopus histone), 4.4 ul of 34 mg/ml chloroamphenicol, 30 ul of 20% glucose, 5 ml of 2X TY, and one colony of transformed cells. Cells were then grown to an optical density of 0.6. The optical density was determined by measuring 1 ml of cells using a UV spectrometer set at 600 nm. The 1 ml samples of cells were then saved. The two 20 ml culture tubes with cells at an OD of 0.6 were split into two different tubes, with each tube containing 2 ml of cells. The protein expression was induced with 5 ul of 0.4 M IPTG, and these cells were incubated for two more hours at 37°C. While the induced cells were incubating, the 1 ml samples of cells at 0.6 OD that were saved were spun at 10,000 rpm for four minutes in a microcentrifuge. Following spinning, the supernatant was disposed and cells were resuspended in 50 ul of 1X SLB and boiled for ten minutes. Samples were then vortexed at maximum speed and preserved either in an ice bucket or at -20°C (if overnight) until ready to load on an 18% SDS Page Gel. The samples were labeled indicating that they were not induced. After induced cells were incubated at two hours, 20 ul of 15 mg/ml Rifampicin (contributed by Luger lab), was added into each tube and incubated for two more hours. Following this incubation, another 1 ml sample was obtained and prepared for loading the same way the uninduced samples were prepared. Prior to loading, samples and marker were boiled for three minutes. Each solution loaded included 2 ul of sample, 5 ul of 4X SLB, and 13 ul of ddH2O.
Future methods and endeavors
Yeast histones (wild and mutants) will be purified using gel filtration and HPLC/ion exchange chromatography, as described in "Reconstitution of Nucleosome Core Particles from Recombinant Histones and DNA" by P. N Dyer et al (2004). Each octamer will then formed using the Histone Octamer Refolding protocol (Dyer et al, 2004). To form the nucleosome, yNAP1 and DNA will be added, and then the nucleosomes will be modified with the photoreactive agent Azido Phenacyl Bromide (APB) along with UV radiation using the protocols in "Site Directed Histone DNA Contact Mapping for Analysis of Nucleosome Dynamics" by Kassabov and Bartholomew (2004). Bartholomew and Kassabov developed these methods so that the site of APB was on the mutant histone, specifically on the cysteine. After UV radiation using a UV transilluminator, APB would react and cross-link the core histone protein with the neighboring nucleosomal DNA. Base NaOH was then added to create a break in the DNA at the site of cross-linking, and the octamer was removed. The DNA break would then be mapped using Taq polymerase primer extension, and the products resolved by running on a sequencing gel (Bartholomew and Kassabov, 2004). This procedure allowed the mapping of the nucleosome locations before and after remodeling.
Results

Figure 6. Sequenced yH4 mutant. Results of sequencing the yH4 after PCR and plasmid isolation. Notice the codon and amino acid change on the 48th base pair (the 47th when methionine on 1st bp is removed); the amino acid has been changed from a serine to a cysteine. Mutation was successful.
The two histone mutations, yH4 S47C and yH2B Q59C, were both generated successfully (Figure 6 and 7). The optimal PCR parameters were found for carrying out these mutations.
Doing a 1% agarose gel for both mutated plasmids with the standard provided the concentration of the yH2B mutant as 20 ng/ul and the yH4 mutant as 100 ng/ul (Figures 4 and 5).

Figure 7. Sequenced yH2B mutant. Results of sequencing the yH2B after PCR and plasmid isolation. Notice the codon and amino acid change on the 60th base pair (59th when methionine on 1st bp is removed); the amino acid has been changed from a glutamine to a cysteine. Mutation was successful.
The gel using the Luger protocol showed expression of yH2B wild, yH2B mutant, and xH2B wild at two hours after induction (Figures 8 and 9). However, the yH4 mutant could not be expressed. The two major goals of this experiment, histone mutation and expression, were accomplished for the mutant yH2B, but not for the expression of mutant yH4.
Discussion

Figure 8. SDS Page gel for expression of xH2B. The expression of the positive control xH2B was successful. M represents the SDS PAGE Biorad marker used. The thick band in B, C, and D represent the xH2B protein expressed by the BL21 (DE3) Codon Plus Competent Cells. Two samples grown in 2 ml of cells that were expressing xH2B were used on the gel. A and B represent cells prior to induction. and C and D represent cells two hours after induction. The well between C and D was an overlap of well D, and is ignored.
Designing primers and using the optimal PCR parameters and the Stratagene Quickchange® Site Directed Mutagenesis Kit (2003) led to successfully mutating yH4 and yH2B to code for the amino acid cysteine. On yH4 the amino acid was changed from a serine to a cysteine on the 48th base pair. On the yH2B sequence, glutamine was changed to a cysteine on the 60th base pair.
Histone expression was a more difficult task. On some attempts, errors were made during histone expression. In previous SDS PAGE gels that were made, bands of protein were present, but not sharp enough. Other times, the gel ran strangely, so the power supply settings were adjusted until the gel ran normally. The expression of yH2B wild, yH2B mutant, and xH2B was successful. The positive control xH2B, YH2B mutant, and YH2B wild type were both expressed two hours after induction; this is not surprising since they differ only in a point mutation on one amino acid. Using Rifampicin aided in the expression, unlike other protocols that did not include the addition of this antibiotic. Expression of yH4 wild was attempted, but this attempt was unsuccessful due to general difficulty in expressing H4 histones. The experiment would have been more successful if yH4 mutant was expressed.
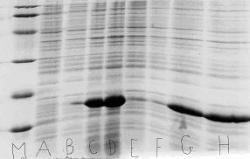
Figure 9. SDS PAGE gel for expression of yH2B wild and yH2B mutant. The histones yH2B wild and yH2B mutant were expressed successfully two hours after induction. M represents the SDS PAGE Biorad marker used. Two samples grown in 2 ml of cells that were expressing yH2B mutant or yH2B wild were used on the gel. A and B represent yH2B wild prior to induction. C and D represent yH2B wild two hours after induction. The bands of yH2B wild protein are very prominent in C and D. E and F represent the cells transformed with yH2B mutant prior to induction, and G and H represent those cells two hours after induction. Note that G and H present a sharp band of yH2B mutant protein.
Two octamers, one containing H4 S47C and the other containing H2B Q59C, will be formed and used in cross-linking experiments with APB. The mapping of the nucleosomes will provide a clearer picture of the location of the nucleosomes on the PHO5 promoter. It will also provide a better understanding of the mechanism of nucleosome remodeling and positioning, since knowledge on this complex is still very limited. If the final result gives researchers a clear picture of remodeling, it will prove that Bartholomew and Kassabov's method of using a photoreactive agent and crosslinking might be the ideal method to map nucleosomes.
Acknowledgements
I am especially grateful to the Luger lab for providing materials and a developed
protocol for the histone expression. I would like to thank the Biochemistry REU program at Colorado State University and the National Science Foundation for giving me this opportunity to excel and discover the endless possibilities in research. In addition, I would like to thank the Laybourn lab, and especially Dr. Paul Laybourn and Rebecca Hart for their guidance, patience, and support. Lastly, I would like to thank my family, friends, and my professors and mentors at Smith College.
References
Bartholomew, B and SR Kassabov (2004) Site Directed Histone-DNA Contact Mapping for Analysis of Nucleosome Dynamics. Methods Enzymol. 375:193-210.
BL21-CodonPlus® Competent Cell Instruction Manual (2003) Stratagene.
Chromosome Organization: Nucleosomes. (2004) What is Life Website.
Dyer, N. P., R. Edayathumangalam, C.L. White, Y. Bao, S. Chakravarthy, U. M.
Muthurajan and K. Lugar. 2004. Reconstitution of Nucleosome Core Particles from
Recombinant Histones and DNA. Methods of Enzymology 375: 24- 37.
Heller, C et al. (2001). Life the Science of Biology. Sunderland, MA: Sinauer.
Horton HR et al. (2002) Principles of Biochemistry. Prentice Hall: New York.
Kassabov, R. S., B. Zhang, J. Persinger and B. Bartholomew. 2003. SWI/SNF Unwraps, Slides and Rewraps the Nucleosome. Molecular Cell 11: 391-403.
Post-translational Protein Modification. National Cancer Institute (2004)
http://www3.cancer.gov/prevention/ptm/summary.html.
QIA prep® Miniprep Handbook (2002) Quiagen.
Quickchange® Site-Directed Mutagenesis Kit Instruction Manual. (2003) Stratagene.
Terrell RA et al. (2002) Reconstitution of Nucleosome Positioning, Remodeling, Histone Acetylation, and Transcriptional Activation on the PHO5 Promoter. Journal of Biological Chemistry 277: 31038-31047.
White, LC et al. (2001) Structure of the yeast nucleosome core particle reveals fundamental changes in internucleosome interactions. EMBO Journal 20: 5207-5218.
Wongwisansri, S (2003) In vivo and in vitro studies of the Role of Chromatin and PHO5 regulation..Colorado State University, Ph.D dissertion: 1-9.
Wongwisansri, S. and P.J. Laybourn (2004) Reconstitution of Yeast Chromatin
Using yNap1p. Methods in Enzymology 375:103-118.