

Author: Sravisht Iyer
Institution: Johns Hopkins University
Date: November 2004
Take anything you learned about genetics in high school and throw it out the door. Incredibly, we may have to do so as we come to understand the prion theory of disease. Suggested to explain Mad Cow and other related diseases, prions are an earth-shaking idea, casting doubt on the status of DNA and RNA as the molecules of life. Not a bacteria, not a fungi, not a virus, but a protein, the prion is a novel infectious agent. The theory that a protein, a non-living object, can propagate disease has become a lightning-rod for controversy and has made the prion hypothesis one of the most hotly contested issues in molecular biology.
Mad cows, shedding sheep, and the birth of the prion theory

Holes in the brain of a sheep infected by scrapie. Groups of PrPSC come together to form rods. Source: Center for Animal Health and Productivity.
The prion theory of disease was created to explain Transmissible Spongiform Encephalopathies (TSEs), a group of fatal diseases that, as they progress, overwhelm neural cells and riddle the brain with sponge-like holes.
TSEs have been found in various species, including sheep, cows, elk and human beings. The disease was first observed by English shepherds in the 18th century when their sheep acted abnormally, rubbing up against any object they could find scraping off most of their skin and wool in the process. The disease, appropriately named scrapies, was devastating, often requiring the sacrifice of entire herds.
Human variants of TSEs are extremely rare, limited mainly to a neurodegenerative disease called Kuru found in cannibalistic Pacific Islanders, and Creutzfeldt-Jakob Disease (CJD), a disorder diagnosed in one person per million. Both these diseases share common symptoms, including gait disorders, jerky movements, and dementia that lead to death months after the first appearance of symptoms.
Throughout the 1970s and much of the 1980s, the prevailing view was that TSEs were caused by a "slow virus," a virus that had a long incubation period. No such virus, however, had ever been purified from the brains of animals that had died from the disease.
When Stanley B. Prusiner, a researcher at the University of California at San Francisco, obtained a pure sample of infectious material, he began working to identify the disease causing agent. When Prusiner added enzymes that destroyed DNA and RNA, he found no change in infectivity. Adding protein-neutralizing enzymes to this cocktail, however, caused a sharp drop in infectivity. This observation set the stage for a theory of disease transmission that Prusiner admitted was "heretical" when he suggested it. In a 1982 Science paper, Prusiner introduced the world to the idea of a proteinaceous infectious particle or "prion" as he dubbed it.
Transformers! Robots in disguise: how prions infect the body
The idea that a protein alone could be an infectious agent remains a contentious issue.
"It has been an extremely difficult scientific question to nail down," said Byron Caughey, a senior investigator National Institutes of Health's Rocky Mountain Laboratories in Hamilton, Montana. "The complete nature of the theory remains unknown and some of the fundamental issues are unresolved."
Researchers believe that at the heart of TSEs is the conversion of a particular protein (known as the Prion-related Protein, or PrP) from a normal (PrPC) to an abnormal, "scrapie-cell" (PrPSC), shape. Both forms of the protein consist of the same building blocks but different final products just like the Transformers toys that could assume two shapes, one benign and the other aggressive. In the prion theory of disease, a PrPSC recruits and re-shapes a PrPC to match its own form, overwhelming neural cells that eventually explode and expose neighboring cells to more PrPSC. As more cells die of infection, they leave behind the spongy, holey brain that is a hallmark of TSEs.
Dr. Jekyll to Mr. Hyde: How PrPs transform
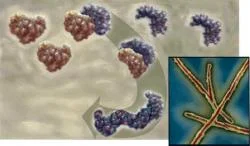
Conversion of PrPC (red) to PrPSC (blue). Groups of PrPSC come together to form rods. Source: The Nobel Committee for Physiology.
Many lines of evidence support the belief that proteins alone are responsible for the transformation of PrPC to PrPSC. Among the most important are the facts that the gene encoding PrP can be found in many species (including humans) and that mice lacking this gene fail to succumb to the disease. Two significant observations can be made based on this data. Firstly, the fact that the body can produce PrP naturally means that no virus is required to supply the PrPSC that triggers the cascade leading to TSE. Secondly, the fact that PrPSC fails to infect animals lacking normal PrP suggests that the key to the disease is the interaction between the two forms of the protein.
Critics of the prion theory, however, were not satisfied by these experiments, claiming that samples of pure protein had never been shown to cause infection in live animals. The lack of such a finding left the door open for the theory that TSEs were caused by bacteria or viruses. In research published in the July 30, 2004 issue of Science, however, Prusiner's group showed, for the first time, that synthetically created proteins can infect mice.
"A great deal of evidence indicates that prions are composed only of protein," says the lead author of the study, Giuseppe Legname, a researcher working in the Prusiner lab, "but this is the first time that this has been directly shown in mammals. The challenge in the last few years has been to figure out exactly how to demonstrate that prions are made entirely of protein."
When they infected mice with synthetic protein, the researchers observed no signs of sickness for 300 days. They were ready to throw in the towel when, on the 380th day, one of the mice showed signs of sickness. By the 660th day, all infected mice had become ill.
While admitting these results represent a key step, Caughey cautioned that they must be taken with a grain of salt. Noting that the results could be attributed to low-level contamination, Caughey added, "In order to settle the question, they need to have all the controls and generate robust, highly-tittered infectious proteins."
Critics of prion diseases have also proposed their own theory to refute the protein-only view supported by Prusiner's lab. Among the most vociferous critics of prion disease has been Dr. Laura Manuelidis, a neurophysiologist at Yale School of Medicine. She has published several papers indicating an immune response in subjects suffering from CJD. This response, she argues, would not be present if the infectious agent was the body's own protein.
Another researcher who has presented dissenting views is Dr. Frank Bastian of Tulane University in New Orleans. Dr. Bastian found traces of a bacteria called spiroplasma in animals and humans suffering from TSE, and he believes that these bacteria, not aberrant proteins, are causing the disease.
Are we there yet? Questions yet to be answered
Recent work has found some key evidence to support the prion hypothesis. There remain, however, several crucial questions about the prion theory that must be answered before statements about it can be made with any certainty.
How is PrP[SUP]C[/SUP] converted to PrPSC? How do prions reach the brain after entering the body? How does PrPSC lead to neurodegeneration? What is the function of PrP in the body?
With so many key questions yet to be addressed, there is still a long way to go for the prion hypothesis, and controversy is sure to follow every step of the way.
References and Suggested Reading
Guyer RL. Prions: Puzzling Infectious Proteins. NIH - Research in the News. (http://science-education.nih.gov/nihHTML/ose/snapshots/multimedia/ritn/prions/prions1.html)
Taubes G. The Game of the Name is Fame. But is it Science? Discover. December, 1986. Reprinted at: http://www.slate.com/id/2096/sidebar/42786/.
Prusiner SB. (1995).The Prion Diseases. Scientific American. (http://www.sciam.com/article.cfm?articleID=0009FD80-C3C6-1C5A-B882809EC588ED9F&pageNumber=1&catID=2)
Prusiner SB. (1982). Novel proteinaceous infectious particles cause scrapie. Science. 9;216(4542):136-44.
Legname G, Baskakov IV, Nguyen HB, et al. (2004). Synthetic Mammalian Prions. Science. 7;305:673-676.
Johnston N. (2004). Clearing Hurdles: Prions Know How to Do It. The Scientist. 18(11):18. (http://www.the-scientist.com/yr2004/jun/feature_040607.html)
Bastian FO, Foster JW. Spiroplasma SP. (2001). 16S rDNA in Creutzfeldt-Jakob disease and scrapie as shown by PCR AND DNA sequence analysis. J Neuropathol Exp Neurol. 60:613-620.
Aguzzi A, Polymenidou M. (2004). Mammalian Prion Biology: One Century of Evolving Concepts. Cell. 116(2):313-327. (http://www.sciencedirect.com/science/article/B6WSN-4BJK582-H/2/70143387edac888b362f04571b6a7988)