
Authors: Kianna J. Mau & Nafisa M. Jadavji
doi: 10.22186/jyi.33.4.63-70
Gastrointestinal dysfunction has a high prevalence in the preclinical phase of Parkinson’s Disease. This review analyzes recent reports that show abnormalities in the gastrointestinal tract of Parkinson’s patients compared to controls, suggesting that the disease originates in the gut. The enteric nervous system, which is composed of myenteric and submucosal plexuses, is susceptible to degeneration in Parkinson’s disease. Evidence regarding Parkinsonian-related loss of myenteric dopamine neurons in the outer plexus of the enteric
nervous system is currently controversial. The diseased submucosal plexus composition, located in the inner enteric plexus has not yet been derived. The dual-hit hypothesis suggests a submucosal role in disease propagation from the gastrointestinal tract to midbrain regions. Parkinson’s patients have an altered gut microbiota composition and varying bacterial concentrations that are correlated with distinct disease phenotypes. These unique bacteria produce short chain fatty acids, which can permeate across the blood brain barrier and indirectly stimulate reactive microgliosis, thus generating a proinflammatory environment that stimulates α-synuclein aggregation. The abnormal Parkinson’s gut microbiota composition has shown to be sufficient in inducing the diseased state. A few reports suggest that diseased microbiome replacement with healthy microbiota via fecal transplant can improve the disease phenotype. Future therapeutic development should target the gut microbiome and its interaction with the enteric nervous system to provide means for an early diagnosis and possible treatment innovations.
Introduction
Parkinson’s disease (PD) is the second most prevalent neurodegenerative disorder in the elderly population, following Alzheimer’s disease (Lin et al., 2014). PD is a chronic disorder, characterized primarily by motor deficits including resting tremor, rigidity, bradykinesia, and postural instability (Burke & O’Malley, 2013; Choi et al., 2016; Lohr & Miller, 2014; Miller et al., 1999; Taylor et al., 2014). Although dopaminergic atrophy in the substantia nigra pars compacta mediates the presence of these motor deficits, the clinical indicators do not appear until over 70% of dopamine (DA) nerve terminals in the striatum have atrophied, suggesting the presence of compensatory mechanisms (Bezard et al., 2013). In disease propagation, alpha-synuclein proteins bind ubiquitin ligands and accumulate in damaged cells (Rao & Gershon, 2016). Alpha-synuclein aggregation leads to Lewy body formation, the characteristic pathological marker. It is currently unclear whether dopaminergic atrophy leads to alpha-synuclein aggregation or if it is the aggregates that lead to cell death.
Few causative factors have thus far been supported, though some environmental toxins have been shown to cause disease symptomology (Pan-Montojo & Reichmann, 2014). For example, exposure to the herbicide Paraquat can result in dopaminergic degeneration and Lewy body formation in the substantia nigra by generating high levels of oxidative stress (Pan-Montojo & Reichmann, 2014). In addition, the production of the synthetic opioid drug MPPP can generate an accidental compound MPTP (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine), which quickly induces a Parkinsonian state when its toxic metabolite inhibits complex I of the electron transport chain (Pan-Montojo & Reichmann, 2014). The possibility of an endogenous neurotoxic mechanism that was acquired in early life has been contemplated for many years (Gibb & Lees, 1988). It is thought that this potential pathogen is transported from the gastrointestinal (GI) tract to the brain via the vagus nerve over the course of twenty years (Syensson et al., 2015). Svensson and colleagues (2015) examined a cohort of patients who underwent vagotomies. They found that patients who received a truncal vagotomy (i.e., the surgical severance of both vagal trunks) had a lower risk of Parkinson’s disease compared to a matched comparison group from the general population (Syensson et al., 2015). Additionally, the basal ganglia are stable throughout the presymptomatic stage (Bezard et al., 2013). It thus appears that the disease origin lies outside the conventional diseased structure (Bezard et al., 2013).
Prior to subcortical disturbance, insoluble alpha-synuclein aggregates have been found throughout the GI tract, causing disrupted gastric motility in transgenic mice (Rao & Gershon, 2016). In the absence of central nervous system (CNS) pathology, genetic mouse models show an increased GI transit time (i.e., longer duration of ingested substance movement through the GI tract compared to the norm) with decreased colonic motility compared to controls (Rao & Gershon, 2016). Increased GI transit time can result in constipation (Rao & Gershon, 2016). Gastrointestinal deficits are some of the most prevalent non-motor symptoms in PD patients and they have shown to affect more than 65% of patients in various cultures (Bugalho et al.,2016; Cheon et al., 2008). Further, the environmental toxins and genetic mutations that initiate PD progression are also associated with enteric nervous system (ENS) deficits. The ENS controls GI tract behavior through innervations in the submucosal and myenteric plexuses surrounding the gut epithelium (Jenkins & Tortora, 2006; Rao & Gershon, 2016). Activity in the ENS is mediated by autonomic input. Disease indicators within the ENS may precede CNS symptoms and pathology, and thereafter propagate to the midbrain region based on the finding that gastrointestinal deficits are present prior to subcortical degeneration (Rao & Gershon, 2016). ENS manifestations could therefore be useful in the future diagnosis and treatment of PD (Rao & Gershon, 2016).
GI dysfunction has a high prevalence in the preclinical phase of PD. Recent hypotheses suggest PD origination in the gut, rather than the brain (Mukherjee et al., 2016; Sampson et al., 2016). This review assesses recent studies that report alterations in the GI tract of PD patients, and as well will describe future therapeutic development in the ENS and its surrounding structures.
Gut-Brain Axis

Figure 1. Flow diagram outlining how the nervous system interacts with the gastrointestinal tract. The nervous system has two components: central, which includes the brain and spinal cord, and peripheral, which comprises all the nerves that extend throughout the body. The peripheral
nervous system is further divided into somatic and autonomic nervous systems. The autonomic nervous system includes sympathetic and parasympathetic
divisions. The sympathetic division controls the fight or flight response, while the parasympathetic division mediates the rest and digest response. The sympathetic division acts through sympathetic nerves to
inhibit enteric nervous system (ENS) activity. The parasympathetic division acts primarily through the dorsal motor nucleus of the vagus nerve to stimulate ENS activity.
The autonomic nervous system (ANS) is a component of the peripheral nervous system. Both divisions control the ENS, making the ENS the largest component of the ANS (Rao & Gershon, 2016). The dorsal motor nucleus of the vagus nerve is the main excitatory parasympathetic input to the GI tract (Hawkes et al., 2007; Mukherjee et al., 2016). Sympathetic nerves also innervate the GI tract, resulting in inhibitory control (Figure 1) (Hawkes et al., 2007).
The ENS contains over 100 million neurons that are intrinsically arranged into microcircuits, which allow the ENS to control the GI tract behaviour without CNS input (Rao & Gershon, 2016). However, neurotransmitter molecules and signalling pathways allow for communication between the two nervous systems (Rao & Gershon, 2016). As such, CNS disease processes frequently have ENS manifestations. Similarly, diseases that originate in the gut routinely have central features. For example, coeliac disease, a matched systemic autoimmune disorder that is triggered by gluten ingestion, often has neurological manifestations, the most common symptom being ataxia (Hadjivassiliou et al., 2010).
The ENS is susceptible to degeneration in PD (Annerino et al., 2012; Fasano et al., 2015; Singaram et al., 1995). Alpha-synuclein aggregations in Lewy bodies are a characteristic PD pathology marker. Aggregates have traditionally been found in the CNS, although growing evidence supports their presence in the ENS as well (Annerino et al., 2012; Fasano et al., 2015; Singaram et al., 1995). The aggregation distribution pattern is most concentrated in the submandibular salivary gland and lower oesophagus, and becomes progressively less concentrated through the stomach, small intestine, colon, and rectum (Fasano et al., 2015). This pattern appears to follow visceromotor projection neuron innervation (Fasano et al., 2015). These neurons originate in the dorsal motor nucleus of the vagus nerve and innervate the extent of the GI tract (Fasano et al., 2015). Communication via vagus nerve input is dominant in the superior regions, while sympathetic innervation is dominant in the inferior GI regions (Fasano et al., 2015). The vagus nerve could thus potentially be acting as a ‘highway’ between gut and brain PD pathologies. It is thought that the disease spreads in a prion-like fashion from the ENS to the CNS, only infecting dopaminergic cells (Hawkes et al., 2007). The vagus nerve has a direct connection with the medulla, which is inferior to the substantia nigra pars compacta. Since this region is dopamine-rich, most of the disease symptomology and pathology stems from substantia nigral degeneration (Mukherjee et al., 2016).
The neurotoxin, 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) is used to model idiopathic PD (Smeyne & Jacson- Lewis, 2005). The MPP+ metabolite causes dopaminergic atrophy by interfering with complex I of the electron transport chain within mitochondria, thereby thwarting energy production (Smeyne & Jacson-Lewis, 2005). Animal models have shown up to a 40% loss of enteric DA neurons within ten days of MPTP administration (Rao & Gershon, 2016). Thus, CNS PD-indicative pathology is also present in the ENS. In the case of PD, major unanswered question is whether the CNS or ENS pathology developed first.
The dual-hit hypothesis suggests that PD begins in the gut and then propagates to the brain. Accordingly, a pathogen infects the brain via both nasal and gastric routes after nasal secretions, containing this unknown pathogen in the saliva, are swallowed (Mukherjee et al., 2016). This viral, neurotropic pathogen is thought to cross the gut epithelial lining. In theory, the pathogen reaches the preganglionic parasympathetic motor neurons of the vagus nerve via transsynaptic transmission through axons from the submucosal plexus, allowing retrograde transport into the medulla (Mukherjee et al., 2016). The process then has downstream effects that allow propagation from the posterior regions to the anterior regions, ultimately infecting the substantia nigra pas compacta (Mukherjee et al., 2016). It is therefore proposed that a neurotropic pathogen initiates PD and is transported to the substantia nigra via the vagus nerve.
Myenteric and Submucosal Plexuses

Figure 2. Enteric nervous system innervations throughout the gastrointestinal
tract. The submucosal plexus innervates the submucosa, which surrounds the mucosa. The mucosa is a mucous layer that lines the gut epithelium. The mucularis encompasses the myenteric plexus and surrounds the submucosa. The serosa, a layer of muscle, surrounds the two plexuses.
The submucosal plexus innervates the submucosa (Jenkins & Tortora, 2006). Adjacent to the submucosa is the mucularis, a circular layer of muscle, which is innervated by the myenteric plexus (Figure 2) (Jenkins & Tortora, 2006). The two plexuses are interconnected (Rao & Gershon, 2016). They are incorporated into 7.5 metres of bowel in a human adult (Rao & Gershon, 2016). Enteric neurons are unevenly distributed throughout the myenteric and submucosal plexuses (Rao & Gershon, 2016). Irregularity of neuronal density throughout the length of the bowel makes quantification of the total neuronal density via paraffin sectioning highly susceptible to sampling error (Rao & Gershon, 2016). Relatively few neurons exist in any one segment (Rao & Gershon, 2016).
Singaram and colleagues (1995) analyzed colon samples from eleven patients with severe PD as well as multiple non-diseased patients. They found that dopaminergic neurons from the colonic myenteric plexus were significantly reduced in PD patients (Singraram et al., 1995) and are correlated to the presence of Lewy bodies in myenteric neurons (Singraram et al., 1995). These results are controversial to other studies, likely due to the technology advances in recent endeavours (Rao & Gershon, 2016). For example, Annerino and colleagues (2012) found no differences in the total density of myenteric neurons between controls and patients with advanced PD. Although neurons containing tyrosine hydroxylase (TH), a dopamine precursor molecule, are most abundant in the stomach, coexistence of Lewy bodies with TH is rare (Annerino et al., 2012). This suggests that myenteric Lewy body formation is not localized to dopaminergic neurons. However, Lewy body pathology in myenteric neurons is synchronous with parasympathetic input from dorsal motor neurons of the vagus nerve, which is known to modulate ENS activity (Annerino et al., 2012). Thus, the researchers suggest that myenteric neuron loss is likely not a characteristic of PD (Annerino et al., 2012). Further investigation of the myenteric plexus is required to elucidate controversial evidence.
Constipation
Constipation is primarily caused by slowed colonic transit in PD patients (Smeyne & Jackson, 2015). Gastric motility takes twice as long in PD-affected individuals (Smeyne & Jackson, 2015. The slowed process is caused by a motor deficit in the GI tract. Lewy bodies accumulate in vasoactive intestinal peptide (VIP) neurons, which amount to nearly 50% of all myenteric neurons (Annerino et al., 2012; Smeyne & Jackson, 2015; Stirpe et al., 2016). Lewy body-filled VIP interneurons disinhibit motor neurons in the GI tract distal smooth muscles, resulting in reflex relaxation impairment (Stirpe et al., 2016). Constipation appears to be more severe in patients who have had PD for many years (Tan et al., 2016).
Biomarkers for constipation are being considered as a prodromal PD diagnostic marker. This non-motor symptom could allow for early diagnosis up to ten years sooner than classical diagnostic measures (Stirpe et al., 2016). However, there is a 15-20% prevalence of constipation in the general population, of which only 30- 60% are comorbid for both constipation and PD (Postuma & Berg, 2016). Thus, constipation has relatively low prognostic integrity.
Constipation has been found to play a role in negatively affecting the absorption of levodopa (Ogawa et al., 2012; Postuma & Berg, 2016; Stirpe et al., 2016; Tan et al., 2016). L-3,4 dihydroxyphenylalanine (L-dopa or levodopa) is the immediate DA precursor molecule (Bianchine et al., 1971). Since DA levels are depleted in midbrain regions of PD patients, levodopa is administered for subsequent DA synthesis (Bianchine et al., 1971). However, the effects of constipation disrupt oral levodopa absorption, and thus motor fluctuations have been observed (Stirpe et al., 2016). A 2012 case study showcased a 69-year-old man who had both PD and intractable constipation for nine years, and unexpectedly presented with parkinsonism-hyperpyrexia syndrome (PHS) (Ogawa et al., 2012). A sudden cessation of dopaminergic activity within the CNS has been shown to cause PHS (Newman et al., 2009). The man in the case study did not discontinue his medication, alter his dosage, or change prescriptions. His physicians concluded that the sudden onset of PHS was triggered by his constipation (Ogawa et al., 2012). Levodopa is absorbed in the small intestine (Ogawa et al., 2012). As such, ingested levodopa is unable to be absorbed for an acute period due to decreased gastric motility and thus cannot be used in the CNS, which can lead to PHS. Hence, PD patients who are comorbid for constipation can experience worsened Parkinsonian symptoms that fluctuate over time due to levodopa malabsorption.
Small Intestinal Bacterial Overgrowth
Compromised gut motility is thought to lead to small intestinal bacterial overgrowth (SIBO) [24]. The small intestine typically contains over one thousand bacteria cells in healthy adults. If the concentration of bacteria cells exceeds one hundred thousand, SIBO syndrome exists [29]. Anaerobic bacteria have a tendency to metabolize sugar molecules into short-chain fatty acids, carbon dioxide, and hydrogen [30]. When SIBO patients exhale, the concentration of hydrogen in their breath is elevated [30]. On the contrary, when healthy humans fast and are at rest, they do not exhale hydrogen as it is produced during anaerobic respiration only [31]. Thus, anaerobic bacteria from the gut generate hydrogen in SIBO syndrome [31].
There are two hydrogen breath tests that can indicate SIBO presence, including the glucose load test and the lactulose test [30]. The glucose load test is more diagnostically accurate and assesses proximal bacterial overgrowth [30]. The lactulose test is less accurate, but can diagnose distal bacterial overgrowth, which is more common [30]. There is currently no universally accepted standard concentration of hydrogen that elicits a definite diagnosis of SIBO [30,32]. Studies report a wide variation of SIBO prevalence in the PD population, ranging from 25.3% to 67% [24,33,34]. Variation between studies could be due to a specific bacterial overgrowth that causes diverse diagnostic interpretation. Methanobrevibacter smithii is a gut bacterium that is present in 15-30% of the general population [32]. It converts four hydrogen atoms into one molecule of methane [32]. People with this bacterium tend to exhale less hydrogen as a result, despite the presence of SIBO, causing a false-negative interpretation.
Once bacterial overgrowth is established, it appears to aid with gut motility by allowing the intestine walls to interact with bacterial metabolites that stimulate movement through the intestines [24]. SIBO is correlated with less severe constipation in PD individuals and some researchers suggest that SIBO could offer some benefit in individuals comorbid for PD and constipation [24].
Alternatively, SIBO induces an inflammatory response in the gut mucosa [24,33]. Mucosal inflammation may interfere with intestinal permeability, allowing toxins to permeate the gut epithelium, leading to microglial activation [20]. SIBO is thus generating a proinflammatory environment, which reduces levodopa absorption and aids alpha-synuclein aggregation in the CNS, leading to worse Parkinsonian symptoms [16,20]. Gut motility is reduced because dopamine levels are low, causing decreased movement through the GI tract. Levodopa malabsorption results from this increased GI transit time. The inflammatory environment mediates alpha-synuclein aggregation via cytokine release and interaction with the proteins. Alpha-synuclein is a regulator of dopamine synthesis through its interaction with TH [35]. However, the overexpression of alpha-synuclein reduces activity in the TH promoter region, leading to an overall reduction in TH levels, thereby further reducing dopamine synthesis [35]. As a result, motor fluctuations have a higher prevalence in diseased individuals who also have SIBO [34]. The bacterial overgrowth is a predictor of worse motor function, independent of disease duration [24].
Gut Microbiota
The diseased gut microbiome is different than that of healthy controls (Scheperjans et al., 2015). Upon PD-derived human gut bacteria implantation, mice develop motor impairment, which is not seen with control-derived human gut microbiota exposure (Sampson et al., 2016). Thus, the composition of gut microbiota in mouse models is sufficient to induce the diseased phenotype. Since these studies have shown that human PD gut bacteria implantation causes Parkinsonian-like phenotypes in mice, it may be important to begin similar studies humans. Sampson and colleagues (2016) suggest that there are certain gut microbiota control pathways that stimulate alpha-synuclein aggregation and inhibit aggregate degradation. Other researchers suggest that reduced beneficial bacterial abundance sanctions an inflammatory outbreak, leading to alpha-synuclein aggregate profusion (Unger et al., 2016). Nonetheless, varying gut bacteria concentrations account for the diverse disease phenotypes (Scheperjans et al., 2015).

Table 1. Variation in the gut composition of control individuals compared to PD patients and the associated symptoms for each microbiota or organic material concentration difference.
1 There are between studies differences in Lactobacillaceae concentration levels in diseased individuals compared to controls.
PD patients have greater Enterobacteriaceae abundance than healthy controls (Table 1) (Forsyth et al., 2011; Unger et al., 2016). The presence of this bacterium is correlated with increased intestinal permeability in PD patients (Forsyth et al., 2011). A compromised intestine wall may expose the ENS to pro-apoptotic factors, possibly driving the PD atrophic sequence (Forsyth et al., 2011). Enterobacteriaceae overabundance is positively correlated with worse postural instability and increased gait difficulty (Mukherjee et al., 2016). Concentration of this bacterial family might be a critical indicator of PD pathology. Some studies report reduced levels of gut Lactobacillaceae in
diseased individuals (Table 1) (Mukherjee et al., 2016). Since vari-ous species promote anti-inflammatory effects, reduced levels re-inforce inflammatory probability (Unger et al., 2016). Lactobacil-laceae appear to modulate the intestinal barrier whereby intestinal inflammation is associated with gut microbiota changes (Mukher-jee et al., 2016; Sampson et al., 2016). The microbiome change might contribute to the misfolding of alpha-synuclein as the proin-flammatory environment mediates protein misfolding via cytokine release and interaction with alpha-synuclein proteins (Sampson et al., 2016). Moreover, reduced Lactobacillaceae concentrations in PD individuals reduce dopamine synthesis, as these species are major producers of the neuroactive compound (Borre et al., 2014).
Other studies report Lactobacillaceae concentration increases. Both Lactobacillaceae abundance and Prevotellaceae reduction are correlated with reduced ghrelin concentration (Table 1) (Scheperians et al., 2015). Ghrelin is a gut hormone that primarily signals hunger, but is also responsible for regulating dopaminergic effects in the nigrostriatal pathway (Bayliss et al., 2016; Scheperians et al., 2015). It is thought to be neuroprotective (Scheperians et al., 2015). Reduced ghrelin concentrations leads to dopamine dysregulation, which facilitates central degeneration (Scheperians et al., 2015). Ghrelin has two forms: des-acylated, which accounts for over 90% of circulating ghrelin, and acylated, which constitutes less than 10% (Bayliss et al., 2016). Chronic des-acylatedghrelin administration increases corticosterone levels (Bayliss et al., 2016). Prolonged exposure to stress is known to have a role in PD development (Bayliss et al., 2016). In contrast, in vivo MPTP exposure shows that acylated ghrelin has neuroprotective effects (Bayliss et al., 2016). The acylated isoform inhibits microglial activation through interaction with the microglial activator MMP-3 in substantia nigral dopamine neurons, preventing successive inflammation (Moon et al., 2009). Accordingly, ghrelin has potential for therapeutic innovation.
Butyrate concentrations are significantly depleted in diseased individuals (Table 1) (Unger et al., 2016). When levels are low, sodium-butyrate concentrations are also low. Sodium-butyrate is a histone deacetylase inhibitor (i.e., a compound that prevents removal of acetyl groups from histone complexes). The compound is dopamine neuroprotective (Unger et al., 2016). A Drosophila PD model experiment demonstrated motor impairment prevention upon sodium-butyrate treatment (St. Laurent et al., 2013). In addition, butyrate beneficially impacts the ENS, promoting colonic contractility by interacting with the colon mucosa (Unger et al., 2016). Thus, reduced butyrate concentrations in PD patients leads to GI dysmotility and indirectly increases dopamine neuron histone deacetylation.
Faecalibacterium prausnitzii is the bacterium that produces butyrate as a metabolite (Unger et al., 2016). Levels are reduced in PD patients (Table 1) (Unger et al., 2016). It is a beneficial gut bacterium that has anti-inflammatory properties (Unger et al., 2016). As such, reduced levels may lead to a compromised intestinal epithelium (Mukheriee et al., 2016; Sampson et al., 2016). Entacapone is a drug that is commonly used to treat PD symptoms (Unger et al., 2016). Its use is negatively correlated with Faecalibacterium prausnitzii abundance, and consequently butyrate abundance (Unger et al., 2016). It is currently unclear whether the diseased state has a pathological Faecalibacterium prausnitzii reduction or whether the medication is causing the decreased abundance.

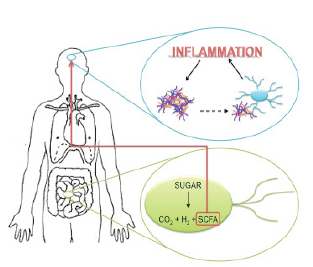
Figure 3. Proposed pathway of disease propagation from the gut to the brain. Anaerobic gut bacteria metabolize sugar into carbon dioxide, hydrogen, and short chain fatty acids (SCFAs). The SCFAs cross the blood brain barrier and indirectly activate microglia. When alpha-synuclein aggregates in the brain come in contact with microglial cells, they also activate them. Microglial hyperactivation leads to inflammation. Proinflammatory environments promote alpha-synuclein aggregation, driving a feedforward inflammatory cascade. (Nygaard, 2010; Sampson et al., 2016)
Mice that are genetically manipulated to overexpress alpha-synuclein show both fine and gross motor deficits, and have GI dysfunction (Sampson et al., 2016). Their gut motility is significantly reduced, resulting in constipation (Sampson et al., 2016). The transgenic mice display low levels of GI microglial activity, increased alpha-synuclein inclusions, and increased motor deficits (Sampson et al., 2016). These symptoms are eliminated in mice whose gut bacteria are diminished by antibiotics (Sampson et al., 2016). However, when the germ-free mice are treated with short chain fatty acids (SCFAs), the diseased state is restored (Sampson et al., 2016). SCFAs are metabolites from the bacterial breakdown of carbohydrates (e.g., butyrate, acetate, propionate) (Borre, et al., 2014; Sampson et al., 2016). When germ-free mice that overexpress alpha-synuclein are treated with SCFAs, their microglia are significantly larger in diameter, and have fewer branches that are shorter in length compared to wildtype mice treated with SCFAs (Sampson et al., 2016). Accordingly, SCFAs potentially mediate gut-brain immune signalling, and their concentration alteration in the diseased state may foster gut-brain dysregulation.
Sampson and colleagues (2016) propose a potential pathway for the pathophysiology of PD from the gut to the brain. Gut microbiota produce SCFAs and the metabolites cross the blood brain barrier and indirectly activate microglia (Sampson et al., 2016). When CNS alpha-synuclein aggregates come in contact with microglial cells, they also activate them (Sampson et al., 2016). Reactive microgliosis involves the release of proinflammatory molecules (Forsyth et al., 2011). Proinflammatory environments promote alpha-synuclein aggregation, driving a feed-forward inflammatory cascade that results in cell death and disease propagation (Figure 3) (Sampson et al., 2016).
Discussion and Future Directions
It is evident from recent studies that the gut is involved in Parkinson’s disease. Constipation is an early non-motor symptom, which has sparked intrigue to analyze the gut-brain axis and moreover the ENS. Reports have shown a strong implication of gut bacteria in disease pathology. Microbiome alterations appear to play a significant role in the disease phenotype. However, the diseased state does not elicit a noteworthy effect on the phenotype of myenteric neurons [21]. Thus, the specific myenteric neuron subtype is not correlated with pathology susceptibility. In addition, the exact compartment of alpha-synuclein aggregation commencement is unknown [21]. Annerino and colleagues (2012) examined the myenteric plexus in a subset of patients with advanced PD and found aggregation throughout the entirety of the GI tract. Future endeavours should use a cohort with a wide range of diseased states, including those who are likely to develop PD. A timeline for gut PD pathology development has not yet been produced. Furthermore, the timing of ENS inception compared with CNS propagation has not been discovered. Such evidence would be sufficient in answering the sought after answer regarding whether the gut or brain is affected first.
The dual-hit hypothesis suggests that a viral neurotropic pathogen initiates Parkinsonian degeneration after entering the body through nasal mucous [15]. The secretions drain into the stomach and are thought to penetrate the epithelial lining [15]. Submucosal axons are believed to mediate pathogen transmission to the vagus nerve [15]. The neurotoxin is assumed to propagate into the medulla and ultimately infect the midbrain region [15]. Although this hypothesis has not yet been evidentially supported, it could be the critical connection between gut and brain interdependence in PD pathology. To our knowledge, the diseased submucosal plexus has yet to be examined. Since the dual-hit hypothesis suggests a role of submucosal axons and since pathology in myenteric neurons is controversial [19,21], there is a demand to expose the accurate function of the submucosal plexus in disease propagation.
A prominent barrier in quantifying and analyzing gut dopaminergic cells lies in the method of identification. TH immunoreactivity is prominently used to detect dopamine neurons [8]. The problem with using this method in the gut is that TH coexists in dopamine and norepinephrine neurons [8]. The gut is innervated by large amounts of noradrenergic sympathetic axons that extend from the extrinsic prevertebral ganglia to the bowel [8]. Staining for both TH and dopamine β-hydroxylase, an enzyme that converts dopamine into norepinephrine would thus be advantageous. Future research must address this issue and consider other identification techniques.
Reduced Faecalibacterium prausnitzii levels result in low butyrate concentrations in PD individuals [37]. Butyrate concentrations can be measured from fecal samples [37], which is a non-invasive tool that could potentially be used as a diagnostic marker. However, it is currently unclear whether the reduced bacterial levels are pathologically derived or whether they are triggered by Entacapone use [37]. In pursuance of potentially using butyrate quantification as a diagnostic strategy, it is first critical to elucidate whether Entacapone is causing the concentration change.
Reduced Prevotellaceae abundance is also observed in PD patients [15,36,37,43,44], though the observation is not exclusive to PD as it is also present in autism and type I diabetes [36]. Fecal samples could however be used to exclude a PD diagnosis [36]. If fecal Prevotellaceae levels are high, PD is likely not diagnostically appropriate. However, if levels are reduced, PD remains a diagnostic candidate. Since this method will not definitively reveal a PD diagnosis, further strategies will need to be implemented.
Although partially invasive, a colon biopsy, whereby researchers are looking for alpha-synuclein aggregates, has potential as a diagnostic approach [23]. The ENS is certainly more accessible than the brain. However, current microbiome knowledge and imaging is not yet sufficiently advanced to use this strategy. For instance, current studies have not yet evaluated the differences in gut microbiomes of individuals with PD, constipation, and those who are comorbid for the two. This comparison would be useful in identifying specific bacterial concentrations in the diseased gut alone, furthermore having potential to illuminate PD specific biomarkers. Future endeavours should certainly focus on accumulating correlative evidence surrounding this notion.
Not only can the gut microbiome be used to potentially diagnose PD, but moreover it may be informative in devising treatment options. Recent studies suggest the importance of ghrelin investigation in neuroprotective efforts against PD progression [40]. Acylated ghrelin has been administered experimentally with promising results in slowing PD progression [40]. It would thus be valuable to consider the use of acylated ghrelin as a potential therapeutic technique, especially during the early stages of neurodegenerative diseases.
Sampson and colleagues (2016) have shown that the diseased microbiome is sufficient to induce Parkinsonian symptoms. It may alternatively be possible for the removal of the diseased microbiome to also remove disease symptoms. One patient with PD has shown symptom improvement in response to fecal transplant [45]. Further, three patients with multiple sclerosis who underwent a fecal transplant regained walking ability [46]. Gut bacteria, possibly through the normal metabolism of carbohydrates into SCFAs, thus have a strong impact on motor functioning. Future investigation of the interaction between the microbiome and PD pathology in order to develop more effective therapies is encouraged.
REFERENCES
Ananthaswamy, A. (2011) Faecal transplant eases symptoms of Parkinson’s disease. New Sci. 209, 8–9
Annerino, D.M. et al. (2012) Parkinson’s disease is not associated with gastrointestinal myenteric ganglion neuron loss. Acta Neurophathol. 124, 665–680
Aroniadis, O.C. and Brandt, L.J. (2013) Fecal microbiota transplantation: past, present and future. Curr. Opin. Gastroenterol. 29, 79–84
Bayliss, J.A. et al. (2016) Acylated but not des-acyl ghrelin is neuroprotective in an MPTP mouse model of Parkinson’s disease. J. Neurochem. 137, 460–471
Bezard, E. et al. (2003) Presymptomatic compensation in Parkinson’s disease is not dopamine-mediated. Trends Neurosci. 26, 215–221
Bianchine, J.R. et al. (1971) Metabolism and absorption of L-3,4 dihydroxyphenylalanine in patients with Parkinson’s disease. Ann. N. Y. Acad. Sci. 179, 126–139
Borre, Y.E. et al. (2014) The Impact of Microbiota on Brain and Behavior: Mechanisms & Therapeutic Potential. In Microbial Endocrinology: The Microbiota- Gut-Brain Axis in Health and Disease (Lyte, M. and Cryan, J. F., eds), pp. 373–403
Bugalho, P. et al. (2016) Non-Motor symptoms in Portuguese Parkinson’s Disease patients: correlation and impact on Quality of Life and Activities of Daily Living. Sci. Rep. 6, 1–9
Burke, R.E. and O’Malley, K. (2013) Axon degeneration in Parkinson’s disease. Exp. Neurol. 246, 72–83
Cheon, S.-M. et al. (2008) Nonmotor symptoms of Parkinson’s disease: prevalence and awareness of patients and families. Parkinsonism Relat. Disord. 14, 286–90
Choi, J. et al. (2016) In Vitro and in Vivo Neuroprotective Effects of Walnut (Juglandis Semen) in Models of Parkinson’s Disease. Int. J. Mol. Sci. 17, 1–17
Donald, I.P. et al. (1992) The Diagnosis of Small Bowel Bacterial Overgrowth in Elderly Patients. J. Am. Geriatr. Soc. 40, 692–696
Eisenmann, A. et al. (2008) Implementation and interpretation of hydrogen breath tests. J. Breath Res 2, 1–9
Fasano, A. et al. (2015) Gastrointestinal dysfunction in Parkinson’s disease. Lancet Neurol. 14, 625–639
Forsyth, C.B. et al. (2011) Increased intestinal permeability correlates with sigmoid mucosa alpha-synuclein staining and endotoxin exposure markers in early Parkinson’s disease. PLoS One 6, e28032
Fung, T.C. et al. (2017) Interactions between the microbiota, immune and nervous systems in health and disease. Nat. Neurosci. 20, 145–155
Ghoshal, U.C. (2011) How to Interpret Hydrogen Breath Tests. J. Neurogastroenterol. Motil. 17, 312–317
Gibb, W.R.G. and Lees, A.J. (1988) The relevance of Lewy body to the pathogenesis of idiopathic Parkinson’s disease. J. Neurol. Neurosurgery, Psychiatry 51, 745–752
Hadjivassiliou, M. et al. (2010) Gluten sensitivity: from gut to brain. Lancet Neurol. 9, 318–330
Hawkes, C.H. et al. (2007) Parkinson’s disease: A dual-hit hypothesis. Neuropathol. Appl. Neurobiol. 33, 599–614
Jenkins, G. and Tortora, G.J. (2006) Figure 23-2. In Anatomy and Physiology: From Science to Life (3rd edn) John Wiley & Sons
Kim, J.-S. and Sung, H.-Y. (2015) Gastrointestinal Autonomic Dysfunction in Patients with Parkinson’s Disease. J Mov Disord 8, 76–82
Klingelhoefer, L. and Reichmann, H. (2015) Pathogenesis of Parkinson disease - the gut-brain axis and environmental factors. Nat. Rev. Neurol. 11, 625–636
St. Laurent, R. et al. (2013) Sodium butyrate improves locomotor impairment and early mortality in a rotenone-induced Drosophila model of Parkinson’s disease. Neuroscience 246, 382–390
Levitt, M.D. (1969) Production and Excretion of Hydrogen Gas in Man. N. Engl. J. Med. 281, 122–127
Lin, S.-C. et al. (2014) In vivo detection of monoaminergic degeneration in early Parkinson disease by 18 F-9-fluoropropyl-( 1)- dihydrotetrabenzazine PET. J. Nucl. Med. 55, 73–79
Lohr, K.M. and Miller, G.W. (2014) VMAT2 and Parkinson’s disease: harnessing the dopamine vesicle. Expert Rev. Neurother. 14, 1115–1117
Miller, G.W. et al. (1999) Immunochemical analysis of vesicular monoamine transporter (VMAT2) protein in Parkinson’s disease. Exp. Neurol. 156, 138–148
Moon, M. et al. (2009) Neuroprotective Effect of Ghrelin in the 1-Methyl-4-Phenyl- 1,2,3,6-Tetrahydropyridine Mouse Model of Parkinson’s Disease by Blocking Microglial Activation. Neurotox Res 15, 332–347
Mukherjee, A. et al. (2016) Gut dysfunction in Parkinson’s disease. World J. Gastroenterol. 22, 5742–5752
Newman, E.J. et al. (2009) The Parkinsonism-Hyperpyrexia Syndrome. Neurocrit. Care 10, 136–140
Niu, X.-L. et al. (2016) Prevalence of small intestinal bacterial overgrowth in Chinese patients with Parkinson’s disease. J. Neural Transm. DOI: 10.1007/ s00702-016-1612-8
Ogawa, E. et al. (2012) Constipation triggered the malignant syndrome in Parkinson’s disease. Neurol. Sci. 33, 347–350
Pan-Montojo, F. and Reichmann, H. (2014) Considerations on the role of environmental toxins in idiopathic Parkinson’s disease pathophysiology. Transl. Neurodegener. 3, 1–13
Postuma, R.B. and Berg, D. (2016) Advances in markers of prodromal Parkinson disease. Nat. Rev. Neurol. 12, 622–634
Rao, M. and Gershon, M.D. (2016) The bowel and beyond: the enteric nervous system in neurological disorders. Nat Rev Gastroenterol Hepatol. 13, 517–528
Sampson, T.R. et al. (2016) Gut Microbiota Regulate Motor Deficits and Neuroinflammation in a Model of Parkinson’s Disease. Cell 167, 1469–1480, e1–e5
Scheperjans, F. et al. (2015) Gut Microbiota Are Related to Parkinson’s Disease and Clinical Phenotype. Mov. Disord. 30, 350–358
Singaram, C. et al. (1995) Dopaminergic defect of enteric nervous system in Parkinson’s disease patients with chronic constipation. Lancet 346, 861–864
Smeyne, R.J. and Jackson-Lewis, V. (2005) The MPTP model of Parkinson’s disease. Mol. Brain Res. 134, 57–66
Stirpe, P. et al. (2016) Constipation: an emerging risk factor for Parkinson’s disease? Eur. J. Neurol. 0, 1–8
Svensson, E. et al. (2015) Vagotomy and Subsequent Risk of Parkinson’s Disease. Ann. Neurol. 78, 522–529
Tan, A.H. et al. (2014) Small intestinal bacterial overgrowth in Parkinson’s disease. Park. Relat. Disord. 20, 535–540
Taylor, T.N. et al. (2014) Reduced vesicular storage of catecholamines causes progressive degeneration in the locus ceruleus. Neuropharmacology 76, 97–105
Unger, M.M. et al. (2016) Short chain fatty acids and gut microbiota differ between patients with Parkinson’s disease and age-matched controls. Park. Relat. Disord. 32, 66–72
Venda, L.L. et al. (2010) α-Synuclein and dopamine at the crossroads of Parkinson’s disease. Trends Neurosci. 33, 559–568