

JENNA MARVIN, REBEKAH SAMUELSON, DYLAN UIBEL, JACOB LAUGHLIN
University of North Alabama, 1 Harrison Plaza, Florence, AL 35632, USA
ABSTRACT
Cystic Fibrosis (CF) is a genetic disease that affects the thickness of digestive fluids, mucus, and sweat, which often leads to obstructions in body organs ducts. The most common CF mutation is the removal of amino acid phenylalanine at position 508 (deltaF508) on the CFTR gene. On CFTR, there are variants of uncertain significance (VUS) as well as classified variations that may cause similarly negative effects. The missense point variations Q493P, W496R, G500D, and Y515C were VUS when this research project began. The aim of this study was to classify them as harmful, neutral, or beneficial.
These variations are non-conservative, meaning that there is a change in the biochemical properties between substituted and original amino acids. A drastic change in the biochemical properties might be detrimental to protein folding and functionality. The variants were assessed through the “sequence-to-structure-to-function” workflow developed by the Prokop lab. Data gathered from several databases were used to determine the variants’ impact on CFTR function by comparing them to all known variants as of October 2019.
Prediction program data was gathered for comparison purposes. Variation analyses included molecular dynamics simulations, CFTR structure analysis, and comparison of the four variations to deltaF508. The amino acids 493, 496, 500, and 515 were shown to fall in highly conserved protein regions under high selection. Therefore, protein functional changes are likely if a substitution was to occur. The insight gained from the analyses may be useful in the identification of other variations and could provide information for classified variants.
AUTHOR SUMMARY
We conducted research to determine effects of variants of uncertain significance (VUS) Q493P, W496R, G500D, and Y515C in the CFTR gene. The CFTR gene contains genetic material to create a membrane protein. Variations of this gene can cause cystic fibrosis. The impacts of Q493P, W496R, G500D, and Y515C were compared to the variant deltaF508 that is known to be associated with cystic fibrosis. We determined the locations of amino acids 493, 496, 500, and 515 in stable and conserved regions on the CFTR protein. Data gathered suggest the amino acid regions have been maintained throughout evolution. Therefore, protein functional changes are likely to be induced if variations occur at these sites. Although these variants were classified during this investigation, they may have similar characteristics to many yet unclassified variants. Classifying variants of uncertain significance would yield more efficient screening methods, treatments, and genetic counseling.
INTRODUCTION
The Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) gene is located on the seventh chromosome. This gene codes for the CFTR protein, a chloride ion and bicarbonate channel (transporter) protein (Sanders et al., 2020). The CFTR protein is responsible for the regulation of epithelial ions (e.g., chloride ions), fluid homeostasis, and water transport (Apweiler et al., 2004). Mutations (variations) of the CFTR gene can cause Cystic Fibrosis (CF), a homozygous recessive disorder. A homozygous recessive genetic disorder occurs when an individual inherits two defective non-dominant alleles, one from each parent. CF affects the thickness of secreted substances such as digestive fluids, mucus, and sweat which often leads to obstructions in body organ ducts (e.g., in the lungs, pancreas, liver, and intestines) (Apweiler et al., 2004). CF manifestation is characterized by chronic bronchopulmonary disease, elevated sweat electrolytes, as well as pancreatic insufficiency (Apweiler et al., 2004). This disorder affects approximately 70,000 patients globally (Sanders et al., 2020). The most common variant of the CFTR gene is deltaF508 (also referred to as Phe508del and ∆F508), which is a loss of function variant caused by a frameshift mutation. Three nitrogenous bases are deleted which results in the elimination of the amino acid phenylalanine at position 508.
On the CFTR gene, there are several classified variants and variants of uncertain significance (VUS) that may cause similar negative effects. VUS are rare variants that are not well classified or defined due to being understudied. Therefore, it is unknown whether they are harmful (also referred to as pathogenic), neutral (have no effect), or beneficial. Classification of VUS would assist doctors and physicians in treating patients more quickly and effectively. Q493P, W496R, G500D, and Y515C are a few of these notable variations (Sanders et al., 2020). These variations are also located in the same protein domain (NBD1) as deltaF508.
Variant Q493P is caused by a nitrogenous base substitution of cytosine for adenine. The amino acid that results from this substitution is proline (P) for glutamine (Q). Glutamine is a polar amino acid while proline is a non-polar amino acid. Polar amino acids are hydrophilic, and non-polar amino acids are hydrophobic. Hydrophilic amino acids tend to position themselves away from the center of the protein structure in an aqueous solution while hydrophobic amino acids tend to position deeper within the protein structure. Position 493 is located on an unstructured portion of the CFTR protein. Unstructured protein regions do not have an established 3D structure under normal physiological conditions (Schlessinger et al., 2007). However, they often assume a different and more regular structure under certain conditions (Schlessinger et al., 2007). Unstructured regions of proteins are also thought to participate in important functional activities such as regulatory processes (Schlessinger et al., 2007).
In a study by Kilinc et al., (2002), A male patient homozygous for the variant Q493P had mild phenotypic characteristics of CF. At the age of four, he had pancreatic sufficiency and no microbial pathogens were present in his lungs. However, his sweat chloride level was 90 milliequivalents (mEq) per liter. Normal sweat chloride levels usually fall between 10-35 mEq per liter.
Variant W496R is caused by the nitrogenous base substitution of cytosine for thymine. The substitution results in the coding of the amino acid arginine (R) instead of tryptophan (W) at position 496. Tryptophan is a nonpolar, hydrophobic amino acid that changes to positively charged (basic) arginine after the substitution. Position 496 is located on an unstructured portion of the CFTR protein. CFTR mutations can be classified into five different classes, depending on their effects on the CFTR protein (Cystic Fibrosis Foundation, 2017). Since the variant W496R is a missense mutation, it is classified as an insufficient protein mutation (Class 5). This mutation results in a reduced amount of normal CFTR protein at the cell surface.
Variant G500D is caused by the nitrogenous base substitution of adenine for guanine. This results in the coding of the amino acid aspartic acid (D) instead of glycine (G) at position 500. Glycine is nonpolar in nature while aspartic acid is a negatively charged (acidic) amino acid. Position 500 is located on an unstructured portion of the CFTR protein. G500D is closely related to G551D because both variants have the same amino acid substitution. G551D is also considered the third most common mutation for CFTR and is defined as pathogenic.
According to Strub and McCray (2020), G551D is a Class 3 mutation which results in defective CFTR channel gating regulations in the plasma membrane since ATP is prevented from binding. Due to G500D’s similarities to G551D, the G500D variation could have similar effects with the reduction of channel activity related to the prohibition of ATP binding.
The variant Y515C is caused by the nitrogenous base substitution of guanine for adenine. The result is the amino acid cysteine (C) being coded for instead of tyrosine (Y). Tyrosine is a polar amino acid while cysteine is a non-polar amino acid. The biochemical properties of this substitution are the same as the substitution Q493P with differing amino acids. Like position 508, position 515 is located on a helical portion of the CFTR protein. A damaging outcome was predicted (5/5) by an in-silico tool (Landrum et al., 2016).
The primary goal of this study was to classify the variants Q493P, W496R, G500D, and Y515C. We predicted that all of these variants would be harmful and have adverse effects on the CFTR protein structure and function. However, towards the end of the research process these variants, along with several other CFTR variants, were classified in the UniProt database. This was mostly likely due to an update in the overall content of the database. The analysis of these variants did not change after they were identified in the UniProt database. Y515C was classified as having a predicted consequence, but the database did not indicate whether the consequence was beneficial or harmful. Q493P, W496R, and G500D were classified as being potentially harmful as they are likely to cause disease. Results obtained from this study were consistent with the updated UniProt classifications. The secondary goal of this study was to compare these variants to the known pathogenic variant deltaF508 as these variants are located in the same protein domain as deltaF508.
METHODS
The variants Q493P, W496R, G500D, and Y515C were examined through the workflow known as “sequence-to-structure-to-function” (Prokop et al., 2017). This was done by assessing the protein structure and analyzing evolutionary sequences. The variants Q493P, W496R, G500D, and Y515C were first identified with the feature viewer for the P13569 (CFTR_HUMAN) entry in the UniProt database (Apweiler et al., 2004). YASARA (Krieger et al. 2009) bioinformatic modeling software was used to model the CFTR protein as well as the variations. The CFTR protein model with 1480 amino acids was generated, followed by embedding it within a lipid membrane, and adding water to each side of the membrane. Variations of the protein were observed by using the swap function in YASARA to replace the amino acids at positions 493, 496, 500, 508, and 515. The CFTR protein model, in the form of a scene file, was provided by Sanders et al. (2020). Slow homology modeling was conducted with the YASARA software to determine conservation among different animal species.
All variants of the CFTR gene (as of October 2019) were compiled from the CFTR1, CFTR2 (Castellani and CFTR2 team., 2013), ClinVar (Landrum et al., 2016), gnomAD (Lek et al., 2016), TOPMed, and COSMIC (Forbes et al., 2011) databases. All variants were accessed with the prediction programs Align-GVGD (Tavtigian et al., 2006), Polyphen2 (Adzhubei et al., 2010), Provean (Choi and Chan, 2015), and SIFT (Ng and Henikoff, 2003). The compiled information on CFTR gene variants was provided by Dr. Jeremy Prokop and his team at the Prokop lab (Michigan State University) in a Microsoft Excel spreadsheet. This spreadsheet also contained statistical analyses of the compiled data. The variants Q493P, W496R, G500D, and Y515C were compared to all ClinVar, gnomAD, and TOPMed missense variants for the gene CFTR. Molecular dynamics simulations (mds) were performed using YASARA software for 20 nanoseconds (ns) on a CFTR protein model that was embedded into a lipid membrane.
RESULTS
CFTR Protein Modeling
The CFTR protein model was generated for all amino acids (1480 in total) followed by embedding within a lipid membrane and water molecules added to each membrane side (Figure 1a, b). Figure 1c provides a clearer view of all the particular amino acid positions important to this study (Q493, W496, G500, F508, and Y515). Positions Q493, W496, G500, and Y515 fall three dimensionally near F508 associated with cystic fibrosis (Figure 1c).
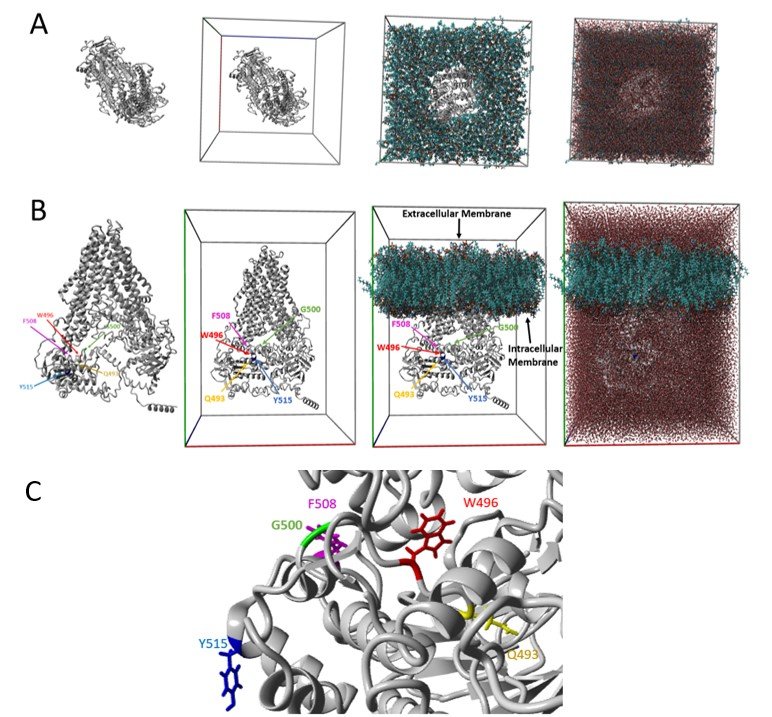
Figure 1. CFTR Structure. (a) Top view of CFTR model, model in simulation box, model embedded into lipid membrane, and water added (left to right). (b) Side view correlating to panel A, with amino acid positions marked. The amino acid position Q493 is highlighted in yellow, W496 is in red, G500 is in green, F508is in magenta, and Y515 is in blue. The intracellular and extracellular membranes are also labeled. Amino acid positions Q493, W496, G500, F508, andY515 are contained on the intracellular side of the membrane. (c) Zoomed in view of CFTR model showing the amino acid position F508 relative toQ493, W496, G500, and Y515. The amino acid position colors correspond to those presented in panel B. All of these amino acid positions are located in the first nucleotide binding domain (NBD1) of the CFTR protein.
Amino Acid Conservation
For each amino acid position, the conservation score was examined for linear motif conservation. This was done by using a 21-codon sliding window additive scoring system. The scores of 10 amino acids were included before and after each position to determine the most conserved linear motifs within this protein. Deep evolutionary analysis of species open reading frames (ORFs) for CFTR revealed the amino acids 493, 496, 500, and 515 to fall in a highly conserved region under very high selection (Figure 2).

Figure 2. Conservation. The chart shows a sliding window calculation for the first1000 amino acids of the CFTR protein (out of 1480 amino acids in total),identifying the most selected and conserved linear motifs. Highlighted in red are the amino acid positions 493, 496, 500, 508, and 515. A zoomed in view of a selected group of amino acid positions ranging from position 490 to 520 are shown in a red box. Amino acid positions highlighted in red represent the five amino acid positions (Q493,W496, G500, F508, and Y515) in which the following variants/variations occur:Q493P, W496R, G500D, and Y515C as well as deltaF508 (associated with Cystic Fibrosis). The numbers above the highlighted sections represent the percent of sequences with synonymous/nonsynonymous variants throughout evolution. Those amino acids positions with a high number of synonymous variants (also referred to as silent mutations) throughout evolution have resulted in a relatively unmodified amino acid sequences, expression of the genes, and phenotypes.
Variant Impact Scoring
In comparison to the predicted impact score produced by averaging all amino acid positions after 508, Q493P, W496R, G500D, and Y515C had similar impact scores (Figure 3). Comparison to the average was used to account for the overall impact of deltaF508, which is a nonsense mutation. Amino acid impact assessments are important for prioritizing CFTR variants with the most potential impact. G500D had the highest impact score while Q493P had the lowest impact score (Figure 3). DeltaF508 had the second highest impact score (Figure 3). The impact scores for Y515C and W496R fell in between those for deltaF508 and Q493P with the impact score for Y515C being the larger value out of the two (Figure 3).
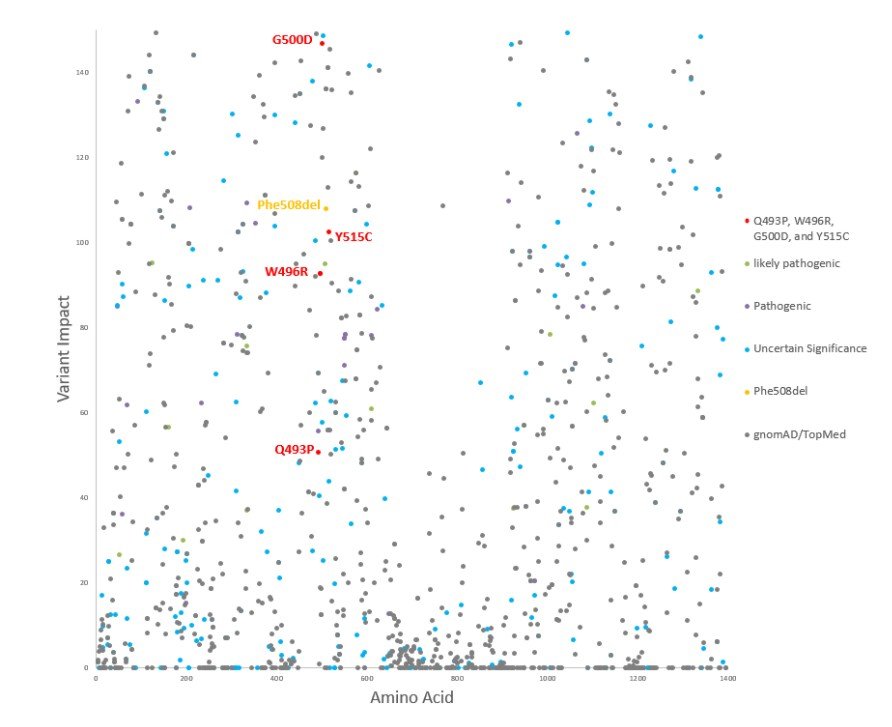
Figure 3. Variant Impact Scatterplot. The scatterplot demonstrates the variant impact scoring for all TOPMed/gnomAD (gray), ClinVar (likely pathogenic-green, pathogenic-magenta, variants of uncertain significance-cyan), and Phe508del (also referred to as deltaF508, yellow) variants for CFTR. Q493P, W496R,G500D, and Y515C variants for CFTR are highlighted in red. SincePhe508del (deltaF508) is a nonsense variant which results in an open reading frame shift, the impact score was found by averaging all amino acid positions after 508. The impact scores for Q493P, W496R, G500D, and Y515C were approximately51, 93, 147, and 103 respectively. The impact score for deltaF508 was approximately 110.
Protein Movement Fluctuation and Deviation
During 20 ns of mds, the CFTR protein reached movement equilibrium which allowed for the calculation of each amino acid’s movement in the simulation. Q493 had the lowest carbon alpha root mean squared fluctuation (RMSF) value while Y515 had the highest carbon alpha RMSF value (Figure 4). F508 had the second highest carbon alpha RMSF (Figure 4). W496R showed the greatest deviation while Y515C showed the least deviation throughout 20 ns of molecular dynamics simulations (Figure 5).

Figure 4. Carbon Alpha RMSF. The carbon alpha root mean squared fluctuation (RMSF) amino acid throughout the 20 nanoseconds of molecular. Amino acid positions Q493, W496, G500, and Y515 are highlighted. The carbon alpha RMSF for Q493, W496, G500, and Y515 were 1.21, 1.32, 1.24, and 2.81 amperes, respectively. The carbon alpha RMSF of F508 was 1.72 amperes.

Figure 5. Global Deviation. The Global Deviation line graph shows the movement of the four variants Q493P (yellow), W496R (red), G500D (green), and Y515C (blue) throughout 20 nanoseconds of molecular dynamics simulations.
CONCLUSIONS
The amino acids 493, 496, 500, and 515 fall in a highly conserved region of the protein under high selection. Therefore, protein functional changes are likely to be induced if a substitution occurred at these amino acid sites. The sequences with synonymous/non-synonymous variants suggest that the amino acid regions have been maintained throughout evolution. Synonymous variants, also known as silent variants, do not change the amino acid.
The variant impacts of W496R and Y515C are similar to the known pathogenic variant deltaF508. The variant impact of G500D exceeded that of deltaF508, and the variant impact of Q493P fell below that of deltaF508. The variants Q493P, W496R, G500D, and Y515C also all have similar scores to other pathogenic CFTR gene variants. Regions Q493, W496, G500, and Y515 demonstrated high stability throughout 20 nanoseconds of simulation according to their low carbon alpha RMSF values. This also suggests protein functional changes if substitutions were induced at these amino acid positions.
Variant W496R had the greatest global deviation, in comparison to the other variants, whereas variant Y515C had the lowest global deviation throughout 20 nanoseconds of molecular dynamics simulations. Variants Q493P and G500D had similar global deviations. The movement patterns suggest that the variants have similar biophysical properties to each other. However, we were unable to compare the variant’s global deviation to that of the wild type (non-mutated protein) due to time constraints. This should be done in future studies of classified variants as well as variants of uncertain significance.
The original goal of this study was to gather information in order to classify the variants Q493P, W496R, G500D, and Y515C. We predicted that all of these variants would be harmful and have negative effects on the structure as well as the function of the CFTR protein. While conducting this research, these four variants were classified in the UniProt database. The results of this study are consistent with the classifications. Y515C was classified as having a predicted consequence. Q493P, W496R, and G500D were classified as likely to cause disease. The insight gained from this study may be useful to the other studies identifying variants of uncertain significance.
There are still many unclassified variants on the CFTR gene as well as other genes that need classification. In addition, those variants that are classified may need further research. Classifying variants of uncertain significance could assist the development of more efficient screening methods and treatments for patients as well as improvements in genetic counseling for patients and their families.
ACKNOWLEDGEMENTS
We thank the following individuals for their assistance. Ms. Michelle Morris of HudsonAlpha provided software access and training. Dr. Jared Painter completed the molecular dynamics simulations for our variants. Dr. Jeremy Prokop provided information about CFTR gene variants, and Dr. Cynthia Stenger mentored us.
REFERENCES
Adzhubei, I.A., Schmidt, S., Peshkin, L., Ramensky, V.E., Gerasimova, A., Bork, P., Kondrashov, A.S., and Sunyaev, S.R. (2010). A method and server for predicting damaging missense mutations, Nat Methods, 7, 248–249, available: https://doi.org/10.1038/nmeth0410-248
Apweiler, R., Bairoch, A., Wu, C.H., Barker, W.C., Boeckmann, B., Ferro, S., Gasteiger, E., Huang, H., Lopez, R., Magrane, M., Martin, M.J., Natale, D.A., O’Donovan, C., Redaschi, N., and Yeh, L.L. (2004). UniProt: The Universal Protein knowledgebase, Nucleic Acids Res, 32(1), D115–D119, available: https://doi.org/10.1093/nar/gkh131
Castellani, C. and CFTR2 team (2013). CFTR2: How will it help care? Paediatr Respir Rev, 14(1), 2–5, available: https://doi.org/10.1016/j.prrv.2013.01.006
Choi, Y. and Chan, A.P. (2015). PROVEAN web server: a tool to predict the functional effect of amino acid substitutions and indels, Bioinformatics, 31, 2745–2747, available: https://doi.org/10.1093/bioinformatics/btv195
Cystic Fibrosis Foundation (2017). Types of CFTR Mutations. Retrieved November 17, 2020, from https://www.cff.org/What-is-CF/Genetics/Types-of-CFTR-Mutations/
Forbes, S.A., Bindal, N., Bamford, S., Cole, C., Kok, C.Y., Beare, D., Jia, M., Shepherd, R., Leung, K., Menzies, A., Teague, J.W., Campbell, P.J., Stratton, M.R., and Futreal, P.A. (2011). COSMIC: mining complete cancer genomes in the catalogue of somatic mutations in cancer, Nucleic Acids Res, 39, D945–D950, available: https://doi.org/10.1093/nar/gkq929
Kilinc, M. O., Ninis, V. N., Dagli, E., Demirkol, M., Özkinay, F., Arikan, Z., Cogulu, Ö., Hüner, G., Karakoc, F., and Tolun, A. (2002). Highest heterogeneity for cystic fibrosis: 36 mutations account for 75 percent of all CF chromosomes in Turkish patients, Am. J. Med. Genet., 113(3), 250–257, available: 10.1002/ajmg.10721
Krieger, E., Joo, K., Lee, J., Lee, J., Raman, S., Thompson, J., Tyka, M., Baker, D., and Karplus, K. (2009). Improving physical realism, stereochemistry, and side-chain accuracy in homology modeling: four approaches that performed well in CASP8, Proteins, 77(S9), 114–122, available: https://doi.org/10.1002/prot.22570
Landrum, M.J., Lee, J.M., Benson, M., Brown, G., Chao, C., Chitipiralla, S., Baoshan, Gu, Hart, J., Hoffman, D., Hoover, J., Jang, W., Katz, K., Ovetsky, M., Riley, G., Sethi, A., Tully, R., Villamarin-Salomon, R., Rubinstein, W., and Maglott, D.R. (2016). ClinVar: public archive of interpretations of clinically relevant variants, Nucleic Acids Res, 44(D1), D862–D868, available: https://doi.org/10.1093/nar/gkv1222
Lek, M., Karczewski, K.J., Minikel, E.V., Samocha, K.E., Banks, E., Fennell, T., O’Donnell-Luria, A.H., Ware, J.S., Hill, A.J., Cummings, B.B., Tukiainen, T., Birnbaum, D.P., Kosmicki, J.A., Duncan, L.E., Estrada, K., Zhao, F., Zou. J., Pierce-Hoffman, E., Berghout, J., Cooper, D.N., Deflaux, N., DePristo, M., Do, R., Flannick, J., Fromer, M., Gauthier, L., Goldstein, J., Gupta, N., Howrigan, D., Kiezun, A., Kurki, M.I., Moonshine, A.L., Natarajan, P., Orozco, L., Peloso, G.M., Poplin, R., Rivas, M.A., Ruano-Rubio, V., Rose, S.A., Ruderfer, D.M., Shakir, K., Stenson, P.D., Stevens, C., Thomas, B.P., Tiao, G., Tusie-Luna, M.T., Weisburd, B., Won, H.H., Yu, D., Altshuler, D.M., Ardissino, D., Boehnke, M., Danesh, J., Donnelly, S., Elosua, R., Florez, J.C., Gabriel, S.B., Getz, G., Glatt, S.J., Hultman, C.M., Kathiresan, S., Laakso, M., McCarroll, S., McCarthy, M.I., McGovern, D., McPherson, R., Neale, B.M., Palotie, A., Purcell, S.M., Saleheen, D., Scharf, J.M., Sklar, P., Sullivan, P.F., Tuomilehto, J., Tsuang, M.T., Watkins, H.C., Wilson, J.G., Daly, M.J., MacArthur, D.G., and Exome Aggregation Consortium. (2016). Analysis of protein coding genetic variation in 60,706 humans, Nature, 536, 285– 291, available: https://doi.org/10.1038/nature19057
Ng, P.C. and Henikoff, S. (2003). SIFT: predicting amino acid changes that affect protein function, Nucleic Acids Res, 31, 3812–3814, available: https://doi.org/10.1093/nar/gkg509
Prokop, J.W., Lazar, J., Crapitto, G., Smith, D.C., Worthey, E.A., and Jacob, H.J. (2017). Molecular modeling in the age of clinical genomics, the enterprise of the next generation, J. Mol. Model, 23, 75, available: https://doi.org/10.1007/s00894-017-3258-3
Sanders, M., Lawlor, J.M.J., Li, X., Schuen, J.N., Millard, S.L., Zhang, X., Buck, L., Grysko, B., Uhl, K.L., Hinds D., Stenger, C.L., Morris M., Lamb, N., Levy, H., Bupp C., and Prokop, J.W. (2020). Genomic, transcriptomic, and protein landscape profile of CFTR and cystic fibrosis, Hum Genet, 140, 423–439, available: https://doi.org/10.1007/s00439-020-02211-w
Schlessinger, A., Punta, M., Rost, B. (2007). Natively unstructured regions in proteins identified from contact predictions, Bioinformatics, 23(18), 2376-2384, available: https://doi.org/10.1093/bioinformatics/btm349
Strub, M.D. and McCray, Jr. P.B. (2020). Transcriptomic and Proteostasis Networks of CFTR and the Development of Small Molecule Modulators for the Treatment of Cystic Fibrosis Lung Disease, Genes, 11(5), 546, available: 10.3390/genes11050546
Tavtigian, S.V., Defenbaugh, A.M., Yin, L., Judkins, T., Scholl, T., Samollow, P.B., Silva, D., Zharkikh, A., and Thomas, A. (2006). Comprehensive statistical study of 452 BRCA1 missense substitutions with classification of eight recurrent substitutions as neutral, J. Med. Genet., 43, 295–305, available: https://doi.org/10.1136/jmg.2005.033878