
Authors: Jason R. Stephenson, Justin B. Morgenthaler, Christopher L. Cooper, Julie A. Stacey, Shani J. Gitter, Jamal Momani, Elizabeth L. Zeitler
Institution: Illinois State University
Date: March 2006
Abstract
The amino acid sequence of the enzyme coproporphyrinogen oxidase (copro'gen oxidase) in the heme biosynthetic pathway is known but its catalytic mechanism has yet to be determined. As a result, it is essential to carry out site-directed mutagenesis on highly conserved amino acids to help determine which amino acids are essential for catalysis. Often, a large number of mutants are created in this fashion, so it is in the researcher's best interest to have a technique to evaluate such mutants efficiently, quickly, and cheaply. In our research lab, we used the His-SelectTM iLAPTM HC Nickel-Coated 96-Well Plate (iLAP plate; Sigma Chemical Company) to evaluate our mutants. We have found that this method was much less labor intensive, more efficient, and significantly cheaper for screening our mutants.
Introduction
Coproporphyrinogen Oxidase (copro'gen oxidase EC 1.3.3.3) is the sixth enzyme in the heme biosynthetic pathway. Heme is an important porphyrin because it carries oxygen through blood as the hemoglobin complex, as well as participates in some enzyme active sites. The authentic substrate for copro'gen oxidase is coproporphyrinogen-III (C-III), which undergoes two sequential oxidative decarboxylations on the A and B rings. Although the amino acid sequence is known, the enzymatic mechanism is not well understood. The aerobic form of the enzyme requires no known cofactor other than molecular oxygen (Medlock and Dailey 1996). This enzymatic activity produces two products: first, a monovinyl product is formed, then the divinyl product. Lash et al. proposed a model (1999) involving sequential oxidative decarboxylations to produce the intermediate monovinyl and divinyl products. This enzyme has been cloned with a 6x-Histidine-tag (6x-His-tag) and can be isolated using nickel affinity chromatography. The 6x-histidines that are part of the primary sequence act as Lewis bases and bind to the Ni2+ resin as reported in the handbook for high-level expression and purification of 6x-His-tagged proteins provided by Qiagen, Inc. Several buffer washes with imidazole are used to compete with the histidine binding and thus disrupt the association of the histidine tag with the Ni2+ resin, thereby eluting the protein. When using site-directed mutagenesis to determine the contribution of various amino acids to catalysis, it is desirable to rapidly and easily screen a large number of mutant enzymes for activity. Therefore, we report here the use of His-SelectTM iLAPTM HC Nickel-Coated 96-Well Plate (iLAP plate; Sigma Chemical Company) as a rapid and selective screening tool for the enzyme copro'gen oxidase. The wells of each iLAP plate were coated with a nickel chelate matrix and lysis components. After cell incubation (3 hr, RT) non-bound materials were removed by a series of washes and the 6x-His-tagged protein eluted with 250 mM imidazole buffer, pH 7.0. The eluates were then evaluated for enzymatic activity, protein concentration (Bradford 1976), and purity by Sodium Dodecyl Sulfate Polyacrylamide Gel Electrophoresis (SDS-PAGE) (Laemmli, 1970). Wild-type and mutant forms of human recombinant 6x-His-tagged copro'gen oxidase were evaluated using the iLAP plates. One of the set of mutants consisted of primary sequence truncations of this enzyme and another set was developed following site directed mutagenesis of single, highly conserved lysine residues. Our experiments with the iLAP plate makes purifying the enzyme and conducting enzyme assays much faster and easier than Ni2+ affinity chromatography.
Materials and Methods
Isolation of Coproporphyrinogen Oxidase Using Two Methods:
Cultures of Escherichia coli containing 6x-His-tagged human wildtype (WT) recombinant or mutant copro'gen oxidase in the vector pET21d were grown at 37° C. The cultures were grown to a selected population density. The population density was measured using a Hewlett Packard UV/VIS spectrophotometer, and it was determined that a value between 0.8-1.0 absorbance units (at 600 nm) indicated sufficient cell growth. Using aliquots from the same cultures, the effectiveness of the iLAP and the nickel affinity chromatography purification methods were directly compared. For the affinity chromatography, E. coli cells harvested from 1 L cultures were lysed using a hydraulic French Press followed by centrifugation at 11,899 x g for 10 minutes. The supernatant was then passed through the Ni2+ affinity column in order to bind the 6x-his-tagged copro'gen oxidase, which was eluted with 250 mM imidazole. An enzyme assay was then performed on the purified copro'gen oxidase (Jones et al. 2003). The second method involved the use of His-SelectTM iLAPTM HC Nickel-Coated 96-Well Plate. Intact E. coli cells (100 microlitersfrom the original 1 liter culture) were added to individual wells of the plate and incubated in the dark for 3-4 hours. Following incubation, liquid was removed from the wells by aspiration and a series of 3 washes were performed using 200 microliters of buffer (0.25 M Tris-HCl, pH 7.0 with 1 mM EDTA) containing 0.05% (v/v) Tween, followed by 2 washes with water. Then 200 microliters of 250 mM imidazole (pH 7) was added to the wells in order to release the bound copro'gen oxidase. The enzyme assay was then performed in each well by adding substrate directly to the wells and, for our control wells, only buffer was used. To measure protein concentration (Bradford 1976) and bound protein by SDS-PAGE (Laemmli 1970), 200 microliters of 250mM imidazole buffer was added to the wells that were not evaluated for enzyme activity, and contents removed for analysis.
Results
Testing Specificity of His-SelectTM iLAPTM HC Nickel-Coated 96-Well Plate

Figure 1. SDS-PAGE (12% gel) analysis of specificity of iLAP Plates. Lane A: Standard copro'gen oxidase. Lane B: iLAP sample of human WT recombinant 6x-His-tagged copro'gen oxidase (20 microliters). Lane C: Wild type E. coli (20 microliters) without WT recombinant human 6x-His-tagged copro'gen oxidase.
In order to test the specificity of the iLAP plate for 6x-His-tagged proteins, two cultures of E. coli were grown to the same approximate cell density. One of the cultures produced only endogenous bacterial wild-type copro'gen oxidase, while the other culture over-expressed the human wild-type recombinant 6x-His-tagged copro'gen oxidase. Both cultures were evaluated using the iLAP plates and samples were taken for SDS-PAGE analysis. Figure 1 is a representative gel showing analysis of proteins eluted from wells. The band in lane B shows that the human WT recombinant 6x-his-tag copro'gen oxidase was bound to the wells of the His-SelectTM iLAPTM HC Nickel-Coated 96-Well Plate, whereas lane C has no band, indicating no bound protein, as expected, indicating that specificity of binding is restricted to proteins only with the 6x-His-tag.
Testing Enzyme Activity of His-SelectTM iLAPTMHC Nickel-Coated 96-Well Plate
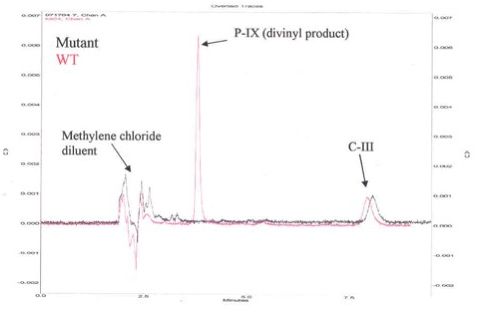
Figure 2. HPLC Chromatogram for Activity of Human Recombinant 6x-His-Tag Copro'gen Oxidase and Representative Mutant Copro'gen Oxidase Enzyme.
Figure 2 is a representative High Pressure Liquid Chromatography (HPLC) chromatogram using a normal phase column (Beckman, Silica, 5μ 4.6 mm x 25 cm) and 35/65 (v/v) ethyl acetate/cyclohexane as solvent. Eluates were evaluated spectrophotometrically at 404 nm. The new peak eluting at 3.8 min indicates active enzyme. To test for activity, the authentic substrate, C-III, was incubated with E. coli preparations containing the human WT recombinant 6x-His-tag copro'gen oxidase either with the nickel affinity purified human enzyme (Figure 3A), or the human enzyme obtained using the iLAP plates (Figure 3B). The data in Figures 3A and 3B show that in both cases, there is more divinyl product produced than monovinyl product with either enzyme preparation, thus both techniques give comparable results, although the magnitude of the responses are different due to lower protein concentration in the iLAP samples. Note, both techniques allow time course studies but the iLAP procedure is less labor intensive while yielding the same trend results.

Figure 3A. C-III Incubation after Copro'gen Oxidase Isolation using Nickel Affinity Chromatography. Each point is the mean of two replicates.

Figure 3B. C-III Incubation after Copro'gen Oxidase Isolation using iLAP Plates. Each point is the mean of two replicates.
Test for Solubility and Activity of Truncated Mutants using His-SelectTM iLAPTM HC Nickel-Coated 96-Well Plate
Five gene truncations were made using the human 6x-His-tagged copro'gen oxidase clone. The shortened constructs contained the following amino acids: 74-354, 90-354, 181-354, 34-177, and 34-233. These mutants were found to all be insoluble and inactive following cell lysis with the French Press and centrifugation. Following incubation in individual iLAP wells, no specifically bound protein was detectable by SDS-PAGE or by protein assay for any of the mutants. Also preparations eluted from the iLAP wells exhibited no detectable enzyme activity for the truncated mutants. Therefore, the iLAP plates provide a rapid screening technique for mutant solubility and activity.
Screening Lysine Mutants of Copro'gen Oxidase using His-SelectTM iLAPTM HC Nickel-Coated 96-Well Plate

Figure 4. SDS-PAGE Analysis of Lysine Mutants and Human WT Enzyme After Purification Using Nickel Affinity Chromatography (Gel 1) or iLAP Plates (Gel 2).

Figure 5. Activity of Wild-Type and Mutant CO after Purification using Nickel Affinity Chromatography (A) and iLAP Plates (B).
To test if lysines 201, 216, 217, and 271 in the primary sequence of human copro'gen oxidase are involved in the active site binding or catalysis, site-directed mutagenesis of these amino acids was performed. Mutant enzyme was either isolated and purified using nickel affinity chromatography, Gel 1, or incubated using SigmaR His-SelectTM iLAPTM HC Nickel-Coated 96-Well plates, Gel 2 (Figure 4) from the same cultures. SDS-PAGE analysis was performed after the purification of human cloned and the four mutant enzymes using the two isolation techniques. It is evident from the gels that both human WT recombinant enzyme and lysine mutant K217A were isolated and purified using both nickel affinity chromatography and the iLAP plates. However, soluble mutant enzyme from K201A, K216A, or K217A constructs was not detected using either method. Human WT recombinant enzyme and the four mutant enzyme preparations from both techniques were also assayed for enzymatic activity. Data are shown in Figure 5. As Figure 5 shows, both isolation techniques indicate the same trends in enzymatic activity. While all mutants exhibited enzyme activity using either technique, only the K217A mutant had substantial activity by either method. Thus, using either technique, we can draw the same conclusion about the general enzymatic ability of the mutants. However, the use of the iLAP plate was much easier and more cost effective since the expensive resin was not needed and the assays could be done using a single iLAP plate.
For insoluble mutant enzymes, growing of the E. coli at lower temperature is often done to help increase the yield of soluble enzyme. Tables 1 and 2 show the percent of specifically bound protein after iLAP plate incubation using E. coli cells containing either WT or lysine mutant recombinants grown at room temperature (Table 2) or 37° C (Table 1). E. coli cells grown at 37° C had a higher percent of specific human recombinant 6x-His-tagged copro'gen oxidase bound protein to the nickel chelate of the iLAP wells than cells at room temperature. Also, at either temperature, there is higher percent specifically bound protein for human WT recombinant and K217A enzymes, which were both soluble, compared with the insoluble K201A, K216A, and K271A mutants.

Table 1. Evaluation of Specifically Bound Protein to iLAP Plates. Specifically Bound Protein using E. coli Cells Grown at 37 degrees C.
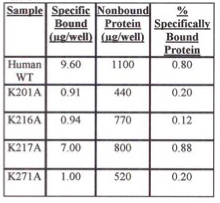
Table 2. Evaluation of Specifically Bound Protein to iLAP Plates. Specifically Bound Protein using E. coli Cells Grown at Room Temperature.
Discussion
The use of either isolation techniques allowed us to isolate, purify, and assess the enzymatic activity of both human WT recombinant enzyme and selected mutants. Both procedures resulted in the same relative conclusions about catalytic ability of the enzymes in question. However, the use of the iLAP plate allowed us to obtain and analyze data much more quickly, easily and cheaply. Affinity chromatography is fairly costly ($687.00/100 ml resin) with 3-5 ml used per column as well as the need for fresh resin for each mutant. This leads to about an approximate cost of $20 for resin per column. It is also rather labor intensive when trying to isolate different mutants. Each iLAP plate contains 96 wells which allows for the maximum number of mutants to be assessed for activity at the same time to be approximately 90 allowing for WT analysis and the other proper controls. A single iLAP plate is also relatively inexpensive costing $65.00 a plate, thus less than 70 cents per well. In addition all the wells need not be used at once allowing for a single plate to be used for more than one experiment, if the plate is stored properly between uses. The iLAP also requires much less buffer (less than 1 ml) compared to the two 50 ml washes of buffer required by the Ni2+ affinity chromatography. A drawback of the iLAP plate method is that it yields a much lower protein concentration than its affinity chromatography counterpart. This reduction in yield was found to have the most impact on the total enzyme activity (product formation) as can be seen in the numeric differences (Figures 3A and 3B) between the iLAP plate and the Ni2+ affinity chromatography preparations. However, this is not a serious problem when only screening for active mutants. This iLAP technology is a highly efficient tool and a reasonable replacement for affinity chromatography especially when considering screening a large number of mutants, or expression of any His-tagged protein. It will be especially useful when screening those mutants that yield insoluble protein. Since it is very difficult to predict insoluble protein ahead of time, researchers will save substantial time and money with this rapid technique.
Acknowledgements
The copro'gen oxidase clone was a generous gift from H. Dailey (University of Georgia, Athens). This work was presented, in part, as a poster at the 24th Midwest Enzyme Conference at the University of Chicago, October 9, 2004.
References
Bradford, M. M. (1976) A Rapid and Sensitive Method for the Quantitation of Microgram Quantities of Protein Utilizing the Principle of Protein-Dye Binding. Analy. Biochem.. 72:248-254.
Jones, M.A., Thientanavanich, P., Anderson, M.D., Lash, T.D. (2003). Comparison of two Assay Methods for Activities of Uroporphyrinogen Decarboxylase and Coproporphyrinogen Oxidase. J. Biochem. Biophys. 55:241-249
Laemmli, U. K. (1970) Cleavage of Structural Proteins During Assembly of the Head of Bacteriophage T4. Nature. 227:680-685.
Lash, T. D., Mani, U. N., Drinan, M. A., Zhen, C., Hall, T., & Jones, M. A. (1998). Normal and Abnormal Heme Biosynthesis. 1. Synthesis and Metabolism of Di- and Monocarboxylic Porphyrinogens Related to Coproporphyrinogen-III and Harderoporphyrinogen: A Model for the Active Site of Coproporphyrinogen Oxidase. J. Org. Chem. 4:464-477.
Medlock, A. E., & Dailey, H. A. (1996) Human Coproporphyrinogen Oxidase is Not a Metalloprotein. J. Biol. Chem. 271,51:32507-32510.